urn:uuid:e7dcc4d6-e1bd-4504-a35e-8b358a6e268cElement Biosciences Bloghttps://www.elementbiosciences.com/assets/img/browser/tile.png?v1https://www.elementbiosciences.com/assets/img/browser/tile-wide.png?v12025-10-06T09:41:51-07:00urn:uuid:edfb028b-00e8-4727-801d-64a496a2ebcdRevealing the immune mysteries of dengue fever with AVITI™2025-10-06T09:41:00-07:002025-10-06T09:41:51-07:00Brian Coullahanbrian.coullahan@elembio.com
A mosquito-borne illness, dengue fever is one of the most widespread infectious diseases worldwide. The World Health Organization estimates that 100-400 million infections occur annually. Dengue severity is linked to the host immune response, particularly B cell-mediated immunity and antibody dynamics.
Historically, it was believed that primary infections were largely mild or asymptomatic while secondary infections caused severe disease due to antibody-dependent enhancement (ADE) and original antigenic sin (OAS). However, recent evidence suggests primary infections may also lead to severe outcomes at comparable rates.
This unique immune dynamic makes dengue not only a global health challenge, but also a complex scientific puzzle. In a recent preprint, Establishment of ‘natural antibodies’ during primary dengue infection, researchers sought to understand the early B cell response using B cell receptor (BCR) sequencing and immunoprofiling of longitudinal samples.
During primary infection, B cells generate antibodies that neutralize the infecting serotype but confer limited cross-protection to other serotypes. In secondary infections, memory B cells generate cross-reactive antibodies, which can sometimes protect but can also worsen disease through ADE and OAS. Dengue also triggers unusual antibody patterns, including low-mutation, broadly reactive antibodies.
Together, these observations highlight additional complexity in immune regulation, with implications for disease progression and vaccine development.
Decoding immunity with BCR sequencing
B cells generate antibodies by recombining variable (V), diversity (D), and joining (J) gene segments in their heavy chain receptor regions. Studying the B cell receptor (BCR) repertoire during infection provides a window into how immunity is built, refined, and sometimes misdirected.
In this study, the authors used heavy-chain BCR sequencing on an AVITI to track the immune repertoire across the course of primary dengue virus (DENV1) infection in a controlled human challenge model. They found that primary infection produces different response patterns across participants—ranging from moderate/delayed activation, to strong plasmablast and IgA/IgM expansion, to an intermediate profile resembling secondary infection, likely due to prior flavivirus exposure.
Ultimately, their work revealed:
Early memory-derived B cell clones—The presence of highly mutated IgA and IgG clones in the acute phase suggests that cross-reactive memory B cells contribute to the early response, even in primary infections.
Emergence of “public” naïve B-cell clones—Multiple participants developed shared BCR signatures, suggesting convergent responses against conserved viral epitopes.
Delayed and atypical maturation—These clones underwent isotype switching (to IgG) without the usual levels of somatic hypermutation, pointing to a T cell–independent pathway of antibody development.
Together, these insights redefine our understanding of how “natural antibodies” arise in dengue infection and highlight why some antibody responses may contribute to protection, while others risk enhancing disease.
Why sequencing technology matters
Capturing this level of immune detail requires high accuracy across the entire variable region. High-quality 300 bp reads are necessary to sequence through the full BCR variable region, ensuring accurate clone identification and lineage tracking. Just as important, high accuracy is essential to detect subtle somatic hypermutations and follow clonal expansion with confidence.
By combining accuracy with read length, AVITI empowers researchers to dissect B cell repertoire dynamics with unprecedented clarity.
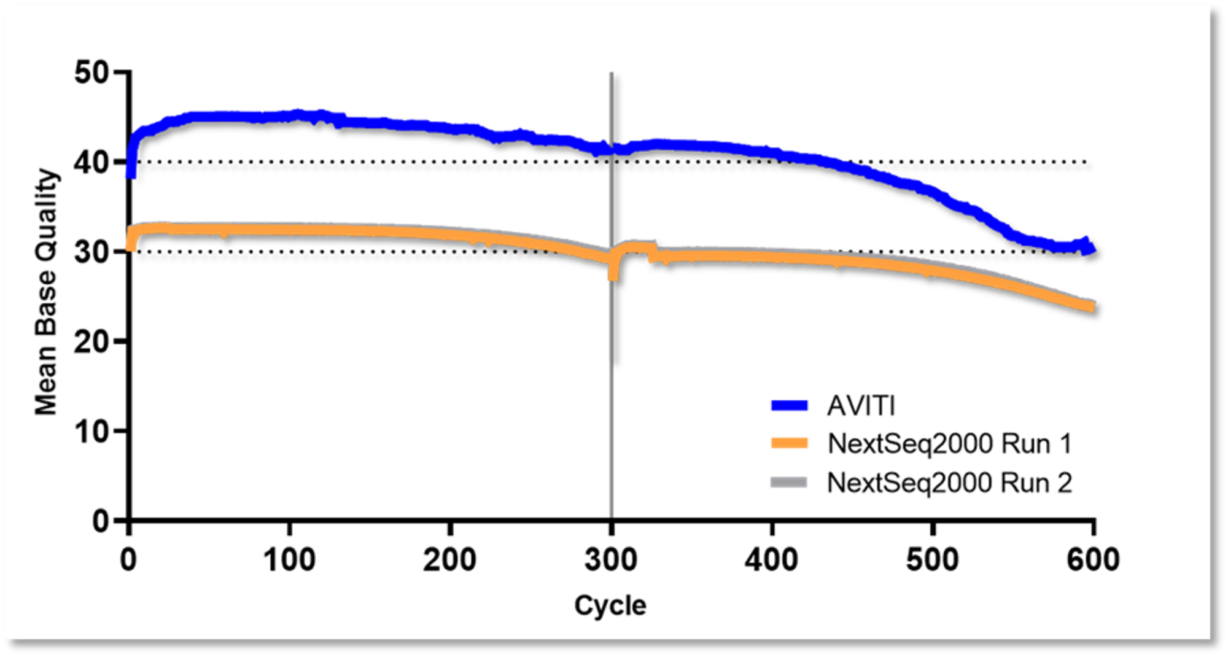
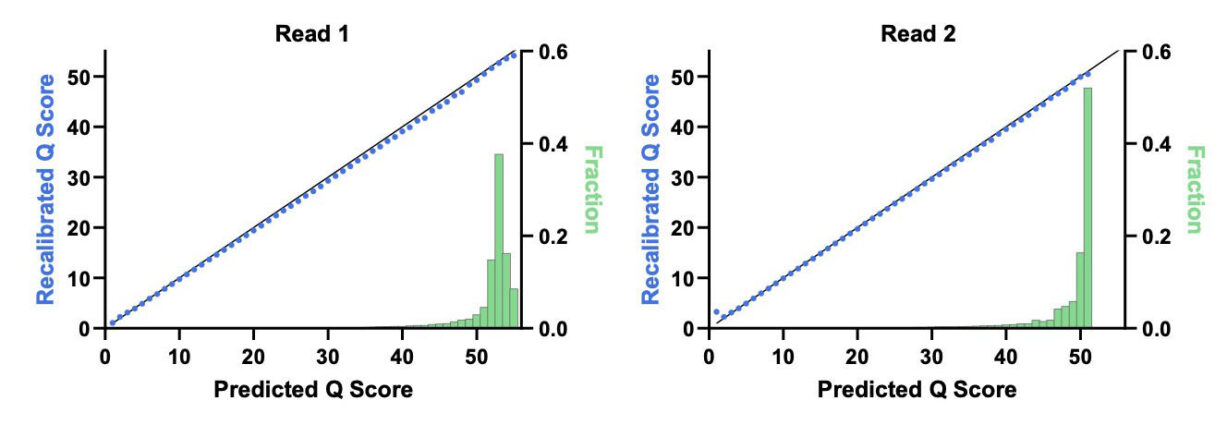
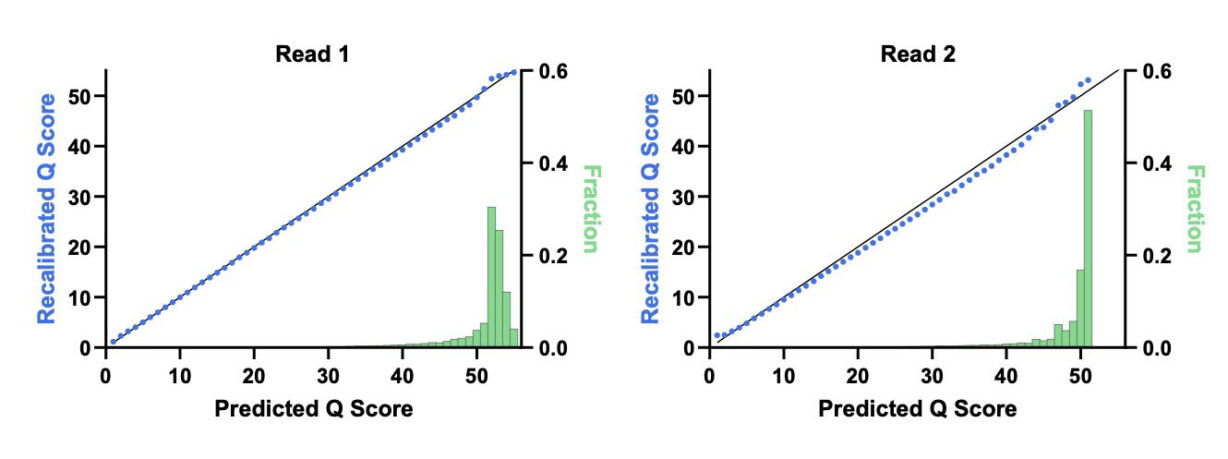
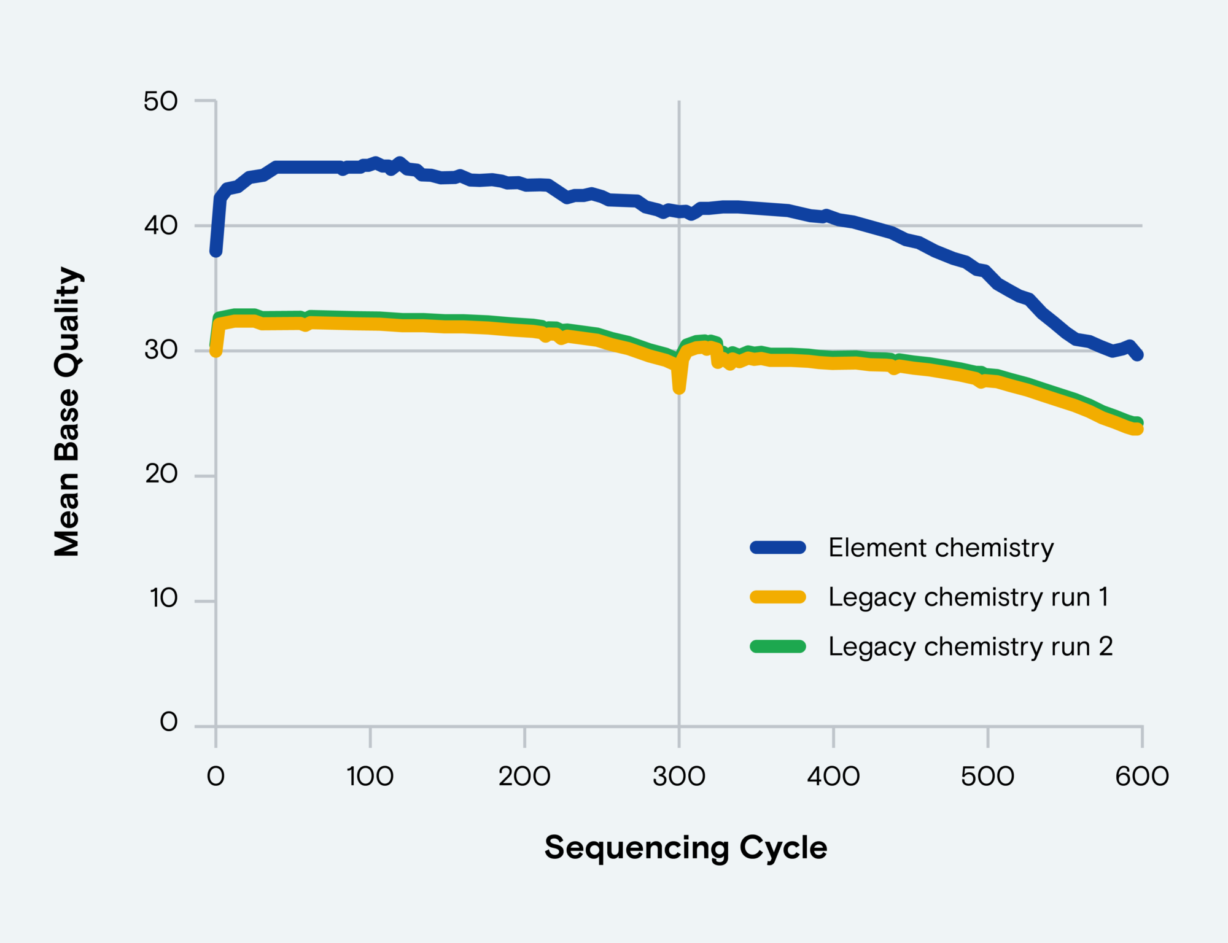
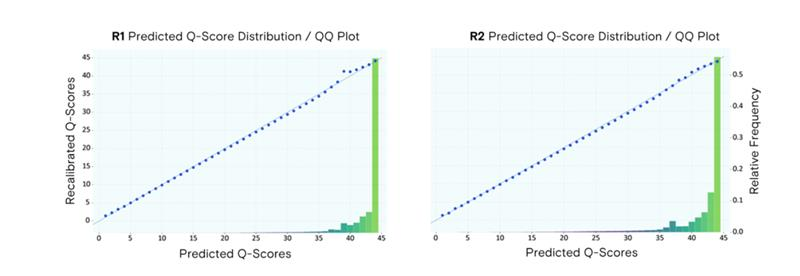
Figure 1. End-to-end read quality over 300 bases. PCR-free Elevate and Illumina fragment libraries for HG001 human control DNA generated using standard protocols. Q values processed using GATK BaseRecalibrator and mean value across replicates plotted across paired reads for both platforms.
Charting a path toward better vaccines and therapies
The fight against dengue underscores a broader truth: our ability to combat infectious diseases depends on our ability to map and understand immune responses at high resolution.
Tools like AVITI will be central to this effort, enabling scientists to discover immune signatures that predict severe disease and identify targets for next-generation dengue vaccines that provide protection without triggering ADE.
Dengue continues to pose a major global threat, but breakthroughs in BCR sequencing are helping decode the immune response that underlies both protection and pathology. With technologies like Avidite Base Chemistry™ researchers can uncover the fine-scale immune dynamics that will ultimately guide us toward safe vaccines and effective treatments.
]]>urn:uuid:d77c355a-304c-4658-8985-e5faf88333f2Why Your Sequencer Shouldn’t Punish You for Wanting More2025-09-24T11:52:00-07:002025-09-24T11:52:41-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
Scientific discovery is built on the pursuit of “more”. Whether it’s more depth to detect rare variants, more throughput to accelerate population-scale projects, or more layers of data to connect genotypes to cellular phenotypes.
Yet in sequencing, getting more often comes at a cost. Historically, researchers who wanted to scale faced a familiar set of barriers: capital-intensive system swaps, constantly increasing consumable costs, or workflow changes that disrupt hard-won efficiencies. These roadblocks don’t just slow labs down, they slow science down.
Rethinking the cost of curiosity
The challenge isn’t just financial. Discovery tools can help shape the pace of research itself and asking bigger questions becomes riskier so scaling a promising program feels harder. Ultimately, opportunities to accelerate discovery can be missed—not because of the science, but because of the available platform or tools.
This model is outdated. As biology discovery tools advance, technology should evolve and expand capabilities in ways that remove barriers, not add them.
Innovation without inflation
At Element, we’re committed to building systems that grow with your research instead of holding you back. Continued innovation and expanded capabilities should be something you can access quickly, without needing to replace your entire platform.
That’s the philosophy behind AVITI24™. It was designed as a high-performance, dual platform that evolves with your science.
Our most recent addition? Through advanced machine learning, AVITI24 now delivers up to 50% more sequencing output compared to AVITI™—up to 1.5 billion reads per flow cell—while lowering the cost per gigabase more than 30%.
Across applications like whole genome sequencing, RNA-seq, single cell RNA-seq, and targeted sequencing, this upgrade translates into tangible benefits: more reads with the same high quality (>90% Q30), more samples per run, and the ability to scale deeper into discovery without added complexity.
And when you’re ready to move beyond sequencing into 5D multiomics, workflows like Teton™ CytoProfiling and Teton Atlas™ with Direct In Sample Sequencing are already built into AVITI24.
For existing AVITI or AVITI LT customers, accessing these expanded capabilities doesn’t require you to buy a new platform. A simple upgrade unlocks AVITI24 performance on the system you already trust with no inflated consumables or system swaps. Just a smarter path forward.
Because your sequencer shouldn’t punish you for wanting more. It should reward your ambition.
]]>urn:uuid:ea772b9c-694b-4615-8130-0cea607971b5Building a future-ready genomics core2025-09-03T11:40:00-07:002025-09-03T11:41:08-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
A conversation with Dr. MeiYeh Jade Lu
For Dr. MeiYeh Jade Lu, NGS Core manager at Academia Sinica, running a high-throughput genomics core isn’t just about staying on top of demand—it’s about delivering the highest data quality for researchers tackling some of biology’s most complex questions.
The Academia Sinica high-throughput genomics core has been operational for over 15 years, supporting cancer research, developmental biology, and genomics projects across a wide range of species. And through it all, one thing hasn’t changed: data quality always comes first.
Why AVITI24™ felt like the right fit
For a facility built on delivering precision to its collaborators, the combination of innovative Avidite Base Chemistry™ and performance metrics made AVITI24 a natural fit when they were selecting a new platform.
“We were looking for a sequencing platform that delivers high accuracy and low background noise without sacrificing efficiency,” Dr. Lu explained. “AVITI24 stood out for its avidite sequencing chemistry, which promised low duplication rates, uniform barcode representation, and critically, a low index hopping rate—all of which are essential for multiplexed studies and high-confidence data.”
Reliable, consistent performance
When it comes to sequencing, consistency and quality are critical for long-term research projects. Uniformity doesn’t just improve data quality but also translates to better cost efficiency. As such, the platform has integrated smoothly into their workflows, with minimal hands-on time and reliable run-to-run consistency.
“AVITI24 has consistently delivered excellent sequencing performance,” she shared. “We’ve been especially impressed by its low index hopping, which helps reduce cross-contamination in multiplexed runs, and the even distribution of reads across barcodes among the same application library type, even with high-plex libraries.”
Finding subtle signals through clean multiplexing
In a recent microbial genomics project, the team needed to multiplex a large number of samples while keeping the per-sample resolution high. Typically, they’d be concerned about clean barcode separation or noise coming into play. However, the AVITI24 has changed how their team approaches these types of studies.
“AVITI24’s low index hopping and clean barcode separation were key to ensuring the integrity of each dataset,” Dr. Lu explained. “This allowed us to detect polymorphisms and/or mutations of low frequencies in the microbial populations, also the 4-color chemistry enables high confident mutation calling that might have otherwise been masked by noise.”
Getting ready for spatial and single cell insights
While the lab currently uses AVITI24 for sequencing projects, Dr. Lu is already planning for what’s next. As they continue to expand their capabilities in larger spatial biology and multiomics studies, the team is exploring how Teton™ CytoProfiling can reveal deeper contextual insights into biology—at a scale and cost-efficiency that will make it accessible to more users at their institution.
“Spatial and single cell multiomics allow us to move beyond bulk profiles and into cellular context and interaction,” she said. “With these tools, we can understand not just which genes are expressed, but where and in what cellular neighborhoods.”
From cancer-immune dynamics to developmental biology to tissue architecture, they’re excited about new ways to integrate cytoprofiling with spatial transcriptomics for a more holistic systems view.
A partnership that goes beyond support
As early adopters of the AVITI24, Dr. Lu appreciates that the Element team has played a key role in helping the lab succeed with transparency and responsiveness.
“Working with Element has been collaborative, proactive, and supportive,” they said. “Their team truly listens, provides rapid technical guidance, and has helped us optimize our applications with confidence. It feels more like a partnership than just a vendor relationship.”
Dr. Lu’s advice for other core labs
As at high-throughput genomics core facility, Dr. Lu, always prioritizes systems that provides both high-quality data and consistent performance to ensure performance across long-term research projects, including data consistency, integration, and batch analysis.
“If you're looking for a short-read platform that combines accuracy, barcode consistency, and low index hopping, AVIT24 is definitely worth exploring. It brings high performance to both standard and advanced applications, and the support team at Element makes onboarding easy.”
And when it comes to future expansion?
“AVITI24 takes that foundation even further, offering the throughput needed for multi-modal spatial and single cell studies with high consistency.”
Download our infographic to learn more about what’s possible with AVITI24, one integrated platform for 5D multiomics and NGS.
]]>urn:uuid:d0ce26e6-f4ad-444d-a6fb-7f8740dadbf6Pangenome-Aware Variant Calling Sets a New Standard2025-07-28T12:51:00-07:002025-07-31T11:52:32-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
Element AVITI™ system delivers the most accurate short-read calls
In genomics, accuracy matters—especially when it comes to identifying genetic variants that impact health, ancestry, or research outcomes. That’s why we’re excited about a recent preprint, Pangenome-aware DeepVariant, from researchers at Google and the UC Santa Cruz Genomics Institute that combines a pangenome reference with advanced deep learning for improvements in variant calling accuracy across multiple platforms. Among them, AVITI sequencing consistently outperformed other technologies, reinforcing its reputation for accuracy and innovation.
Better variant calling with the power of pangenomes
Reference genomes provide a blueprint for read alignment, variant calling, annotation, comparative genomics, and allow us to systematically interpret sequencing data. However, basing variant calling pipelines on a single, linear reference genome introduces bias, especially in regions with high diversity or structural complexity, which can skew downstream data analysis.
To overcome these biases, the Human Pangenome Reference Consortium assembled a pangenome graph to improve read mapping and variant calling. Using this dataset as a foundation, in this study, the authors introduce and assess pangenome-aware DeepVariant, a variant caller using advanced deep learning to identify candidate variant sites and distinguish true variant signals from noise. Overall, the pangenome aware DeepVariant reduced errors and achieved over 20% more accurate variant calling compared to existing methods.
Importantly, this method is generalizable and supports reads from multiple sequencing technologies—and that’s where our AVITI sequencing stands out.
AVITI sequencing data sets the accuracy benchmark
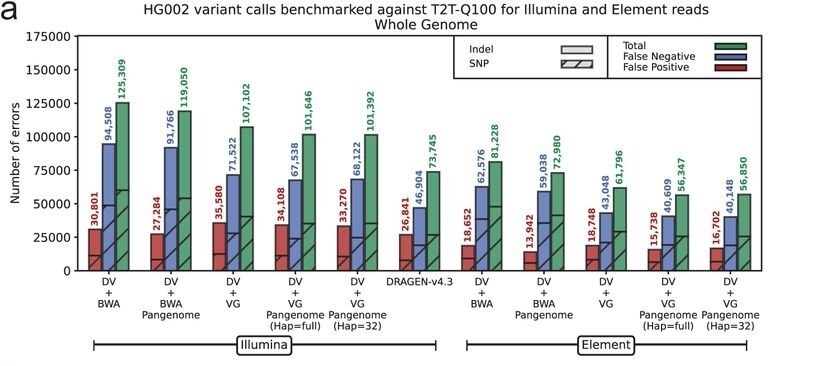
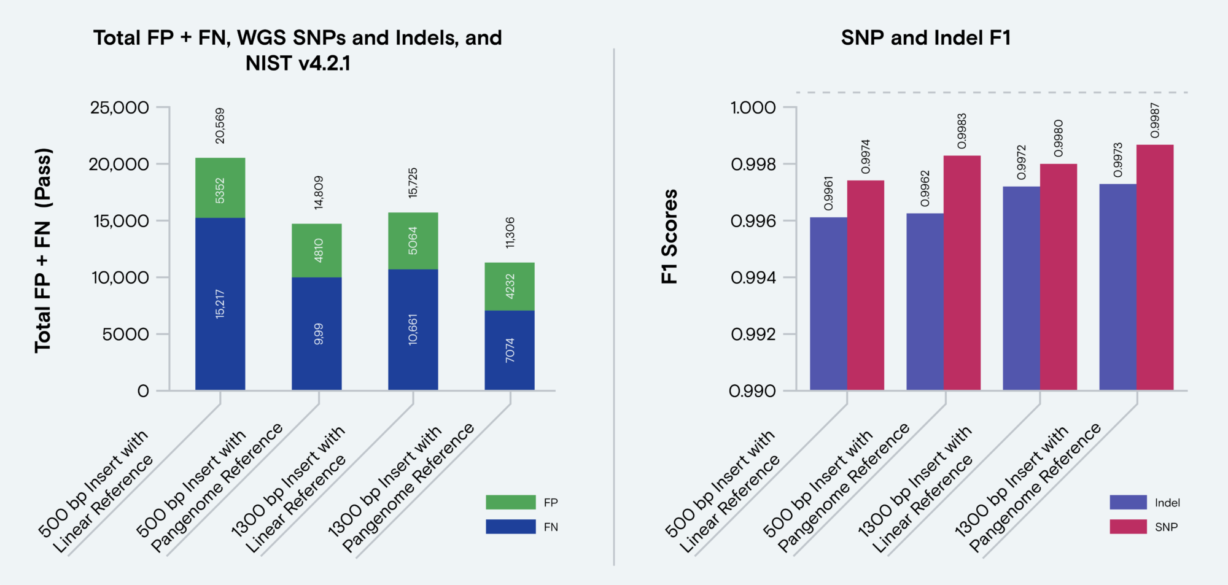
The study evaluated performance of Illumina and Element reads using the benchmark truth set, T2T-Q100-v1.1. Across every mapper and variant caller tested on this truth set, Element AVITI outperformed Illumina in generating accurate variant call sets. The most accurate short-read call set achieved 99.65% precision and 99.1% recall when using AVITI data with vg giraffe mapping and pangenome-aware DeepVariant calling.
Benchmarking Element and Illumina against the T2T-Q100 truth set across T2T high confidence regions across mapping and variant calling tools. Figure from: Pangenome-aware DeepVariant. Mobin Asri, et al.bioRxiv 2025.06.05.657102; doi: https://doi.org/10.1101/2025.06.05.657102. CC BY 4.0.
A powerful validation of our technology, this echoes what we’ve shown before: our avidite base chemistry delivers improved accuracy, especially in difficult genomics regions like homopolymers and short tandem repeats.
The paper also calls attention to how the choice of benchmark matters. The Platinum truth set, built mostly on Illumina data, masked some advantages of AVITI, whereas the T2T-Q100 benchmark highlights AVITI’s accuracy.
Future-proof variant calling for precision genomics
Regardless of input data, pangenome-aware DeepVariant consistently reduced errors, especially in segmental duplications where short reads are prone to mismapping and is designed to keep pace with ever-growing reference datasets.
As the field scales up to richer pangenome references including personalized pangenomes and future HPRC references containing up to 1,000 haplotypes, it’s clear that high-quality sequencing from platforms like the AVITI will play a key role in accurate variant calling.
Final takeaway? This study reaffirms what we’ve believed from the start—high-accuracy sequencing with Element isn’t just better. It’s leading the way.
]]>urn:uuid:fbf64244-ca4d-4220-8998-bd776b4d3841The Future of Multiomic Research in Neuroscience: Exclusive Insights with Dr. Matt Tegtmeyer2025-07-18T13:22:00-07:002025-07-23T17:06:46-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
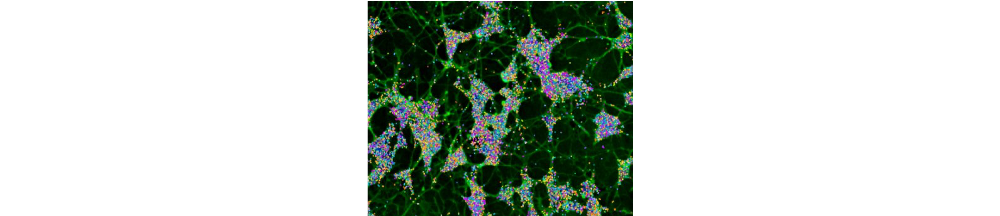
In the rapidly evolving landscape of neuroscience research, the integration of high-dimensional phenotyping with multiomic technologies is transforming our ability to investigate neurodegenerative disease biology. In a conversation with Dr. Matt Tegtmeyer, assistant professor at Purdue University, we discussed his lab's pioneering work in cellular phenotyping and why they’re excited about their early studies with the AVITI24™ platform.
From cell painting to multiomic integration
Dr. Tegtmeyer’s lab specializes in using high-dimensional phenotyping, or phenomics, to understand the mechanisms underlying genetic variants associated with neuropsychiatric and neurodegenerative disorders. His team uses cell painting, a morphological profiling technique, to analyze cellular structures across different cell types and differentiation states.
"We use a mixture of six different stains that measure eight different constituent organelles, including the nucleus, endoplasmic reticulum, mitochondria, actin, Golgi, plasma membrane, and cytoplasmic RNA. Using machine learning, we extract thousands of morphological characteristics from a single cell," Dr. Tegtmeyer explained. He likened this approach to genome-wide expression profiling to get an unbiased global survey of cellular morphology, and emphasized its affordability compared other single cell technologies.
However, one of the major challenges with cell painting is that interpretation of these features requires additional experiments to determine the molecular underpinnings. "Whereas in this case," he noted, referring to the AVITI24 platform, "we could capture RNA, protein, and morphology simultaneously so we can have a comprehensive view of what molecular changes are occurring, and how those might give rise to the morphological changes."
Pilot testing the AVITI24 platform
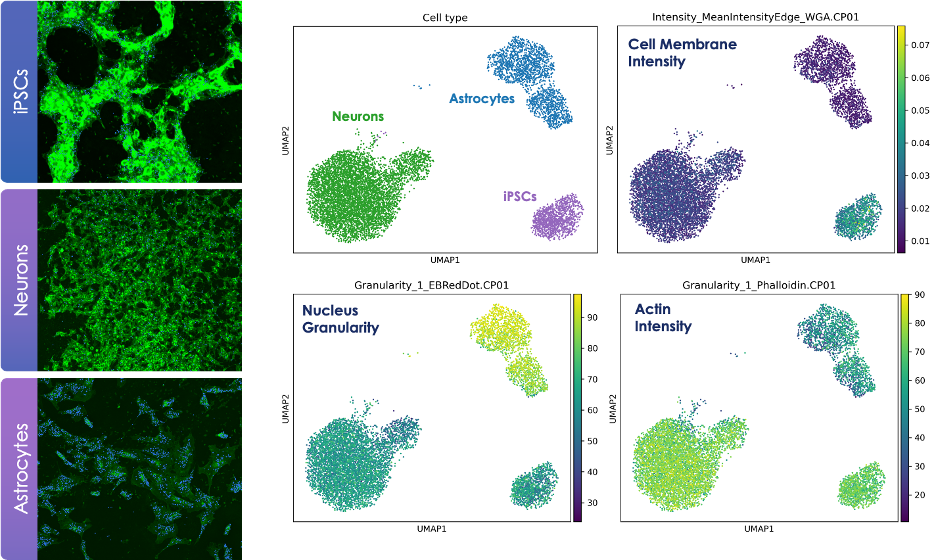
Dr. Tegtmeyer’s lab piloted the AVITI24 platform and our Teton™ neuroscience panel, which allows simultaneous assessment of 350 transcripts, 50 proteins (including phospho-proteins), and 6 cellular morphology features. For these experiments, the team differentiated induced pluripotent stem cells (iPSCs) into astrocytes and excitatory neurons, using a neurogenome overexpression approach, and had their cells analyzed on the AVITI24 as part of our TAP program.
"One of the things I was very interested in was comparing cell painting on the AVITI24 to our standard cell painting methods," Dr. Tegtmeyer shared. "In our recent neural cell painting publication, we achieved 95% accuracy in predicting cell type based on morphology. But with AVITI24, we saw an increase to 99% accuracy using the same logistic regression model. We get better accuracy with the added information of RNA expression.”
Integrating multiomic and morphology features during neuronal iPSC differentiation. Using RNA and protein readouts, the Tegtmeyer lab was able to overlay morphology with cell type clusters to understand how morphology features were changing as cells differentiated.
Linking morphology to disease biology
Beyond cellular classification, the technology is providing valuable insights into disease biology. One of the key findings from their pilot experiments involved the gene CAPN2, which encodes calpain-2, an enzyme linked to Alzheimer’s disease.
"We observed significantly higher CAPN2 expression in astrocytes compared to neurons and iPSCs," Dr. Tegtmeyer revealed. "Since CAPN2 is implicated in the cleavage of GSK3-beta, which exacerbates Tau pathology and neuroinflammation, we’re now investigating how Alzheimer’s-associated variants and environmental toxins impact this relationship. In one single run of an experiment, I can assess the relationship, and I don’t have to rely on puzzling together several rounds and iterations of validations."
Scaling up multiomic research
Dr. Tegtmeyer emphasized the scalability and efficiency of the AVITI24 platform, particularly for high-throughput experiments. "Profiling millions of cells is incredibly expensive and time-consuming with current single cell approaches. But with Teton chemistry, doing a million cells in a run is feasible, and we get data back overnight instead of waiting weeks or months."
This accelerated timeline has profound implications for the field. "Rather than spending months analyzing an experiment and deciding whether the information is useful, we can know in a week and move forward quickly," he added, “By using a system with high dimensional readouts, we can get a better sense of what is causing disease, rather than what is caused by disease.”
Looking to the future
One of the most exciting prospects on the horizon is the potential application of AVITI24 in neuro drug discovery. As his lab continues to push the boundaries of what’s possible with multiomics, Dr. Tegtmeyer is excited about the future.
With the rapid advancements in multiomic technologies, Dr. Tegtmeyer’s work underscores the transformative potential of integrated morphological and molecular profiling. As tools like AVITI24 continue to advance, they promise to accelerate the pace of discovery in neuroscience and beyond.
]]>urn:uuid:592fbc91-99c0-485d-ab09-381bfb9b3f98Chemistry that adapts: Inside the innovation powering AVITI24™2025-06-23T12:30:00-07:002025-07-15T15:58:43-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
Complex biological experiments depend on chemistry that can do more—without demanding more. That’s what Teton™ and Teton Atlas™ deliver: onboard detection chemistries designed to deliver flexible, multimodal readouts across RNA, protein, and cell morphology.
Our latest infographic breaks down this chemistry step-by-step. It’s a visual map of how detection happens at scale, using integrated chemistry that puts flexibility and discovery first.
One platform, multiple modalities
The magic of the detection chemistry starts with its modular design. Barcoded oligo probes and antibody conjugates target RNA and proteins, while a range of dyes and reversible cell paints capture the full cellular landscape. With Teton Atlas for Direct In Sample Sequencing (DISS), you can perform true RNA sequencing in its native context.
Want targeted RNA data? Whole transcriptome coverage? Protein detection? Structural context? You don’t have to choose. Teton Atlas is designed to let you combine them and adjust the panel as your study evolves.
No hand-offs. No extra systems.
Traditional spatial and multiomic workflows often rely on a patchwork of imaging, sequencing, and sample prep systems. With Teton Atlas, it’s all onboard. Every step from probe binding to the final sequencing readout happens on a single platform. That means less hands-on time, fewer potential failure points, and seamless progression from detection to readout.
Visualizing the workflows
The infographic walks through the key detection schemes:
Reversible cell paint: visualization of key morphology features (cell membrane, mitochondria, ER, Golgi, nucleus, and actin) relies on a combination of sequencing readouts and fluorescent stains for improved scalability and increased number of targets.
Barcoding: Automated, onboard protein and RNA detection with our Teton panels using probes that are sequenced over multiple batches for improved scalability.
DISS: Exclusively available with Teton Atlas, DISS enables 3' whole transcriptome and targeted RNA sequencing directly in their cellular context.
Each step is engineered for throughput, fidelity, and adaptability—so your experiments can grow without growing more complicated.
See it for yourself
Whether you're studying tissue architecture, immune environments, or disease progression, the ability to get RNA, protein, and morphological context from a single sample opens doors to more meaningful insights. It’s chemistry that matches your biological complexity with technological elegance.
This infographic reveals how we designed a chemistry engine built to scale with your science. Get the visual breakdown of the onboard, multiplexed detection powering 5D multiomics.
]]>urn:uuid:3958a003-fa40-4ee4-8d80-294b6a8258e3How multiomics can accelerate and support cancer research2025-06-17T14:08:00-07:002025-06-17T14:08:12-07:00Michael Sullivanmichael.sullivan@elembio.com
Unraveling the mysteries of biology often involves identifying its subtleties: what makes our cells tick?
The nature of cells is that they are ever changing. They live. They adapt to their environment. But for decades, scientists have lacked a truly integrated tool for understanding how cell dynamics unfold across multiple biological layers simultaneously, particularly in areas like cancer research.
Our AVITI24™ 5D multiomics system and Teton™ assay change this current paradigm.
A new study from Element scientists
A recent pre-print published by our scientists highlights the power of this system, and its ability to actually capture the effervescent complexity of human cells.
The basis for the paper was a straightforward question: how can we demonstrate the capabilities of our technology in a way that is real and biologically relevant?
To do so, we chose non-small cell lung cancer (NSCLC) drug resistance, a complex problem that typically requires years of painstaking research. An all too common occurrence with NSCLC is that it develops resistance to tyrosine kinase inhibitors (TKIs), limiting the long-term success of these targeted therapies.
The conventional approach to studying this cancer drug resistance involves a tedious, fragmented process. Researchers typically analyze protein or RNA separately, each requiring multiple days of preparation before getting results. Then they repeat this process over and over for different targets, slowly building a picture of what's happening inside cells. It's methodical, but painfully slow.
Accelerating time to insights: From months to weeks
Our Teton assay on the AVITI24 system drastically accelerates this timeline. Instead of measuring RNA, protein, and cellular morphology separately over weeks or months, we capture all three simultaneously in individual cells—going directly from sample to data in a single 24-hour run.
"With AVITI24 and Teton, we were able to cast a really wide net and look broadly at everything that could be impacted within a cell," explained Vivian Dien, Element’s Associate Director and lead scientist on the project. "We can differentiate between the response at a bulk population level, which people typically do, and parse it out to an individual cell level to understand why some cells are responsive while others aren't."
The speed difference here is staggering. Instead of spending years on tedious sample preparation and running separate assays for different pathways and drug combinations, researchers can focus more time on analyzing data and building new hypotheses.
For example, when we identified CDK4/6 as a potential escape route for resistant cells and decided to test combination therapy with palbociclib, the turnaround was incredibly fast.
"From the initial idea to seeing results, it took only about three weeks," Vivian noted. “Our study shows that with this system, what would have previously taken an entire lab’s worth of equipment over many months or even years, can now be done on a single machine in under a single month. It's much more accessible for everyone."
This acceleration is not just about convenience—it's about fundamentally changing how drug research happens. The ability to simultaneously measure hundreds of proteins alongside RNA expression and morphological changes in the same cells opens up entirely new experimental possibilities that simply weren't feasible before.
At Element, we believe modern science deserves modern solutions. Solutions that meet researchers at their pace and resource capacity. As demonstrated in this paper, AVITI24 is that solution. A system working hand in hand with scientists to achieve high quality results faster than ever before.
]]>urn:uuid:cab6d0c5-291c-4681-8dca-6391ef8edaf2Decoding Genetic Variation: De Novo Mutations in Focus2025-06-13T07:19:00-07:002025-06-13T07:19:09-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
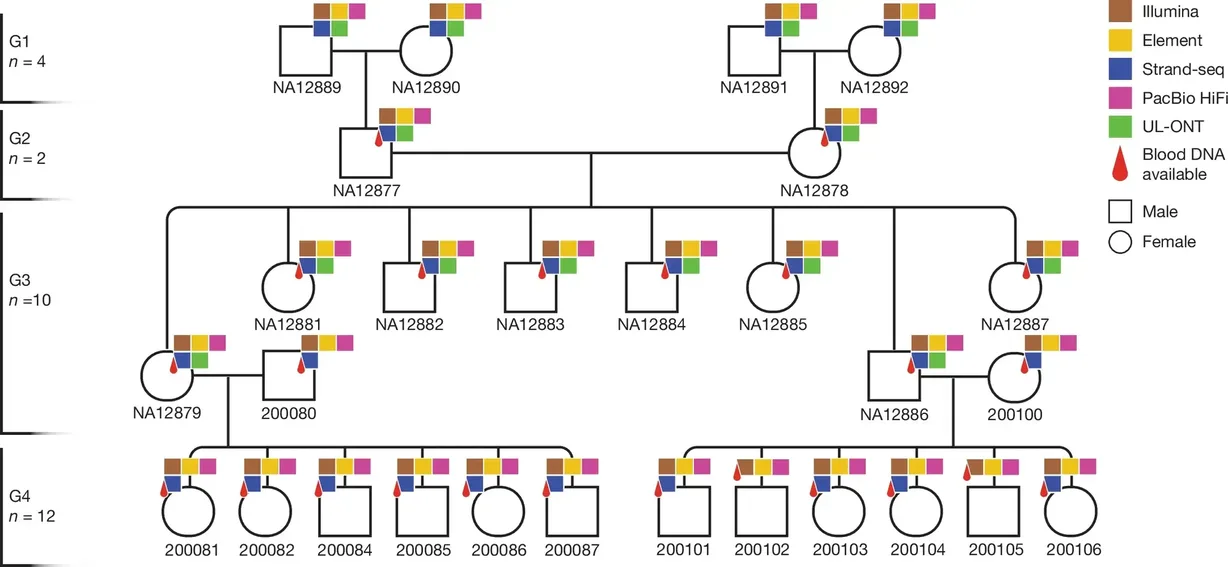
How far can precision sequencing take us in understanding the human genome? The answer becomes clearer with each study that pushes the limits of accuracy, scale, and resolution. One of the latest examples is a deep, four-generation analysis, providing a truth set to uncover critical insights into de novo mutations (DNMs) and their implications for genetic variation.
In this Nature publication, authors from the University of Washington School of Medicine leveraged advanced sequencing technologies, including the Element AVITI™ platform, to phase and assemble over 95% of the diploid genomes of the four-generation, 28-member family (CEPH 1463), with near telomere to telomere coverage.
Study design for sequencing CEPH 1463 pedigree containing four generations and 48 family members. Figure from Porubsky, D., Dashnow, H., Sasani, T.A. et al. Nature (2025). https://doi.org/10.1038/s41586-025-08922-2. CC-BY-4.0
De novo mutations: where variation begins
De novo mutations arise spontaneously, not from inherited DNA, and are a primary source of genetic diversity, a key factor in many rare diseases, and a central focus of rare disease research. Understanding DNMs requires high-resolution phasing, complete diploid assemblies, and the ability to distinguish subtle variation from noise.
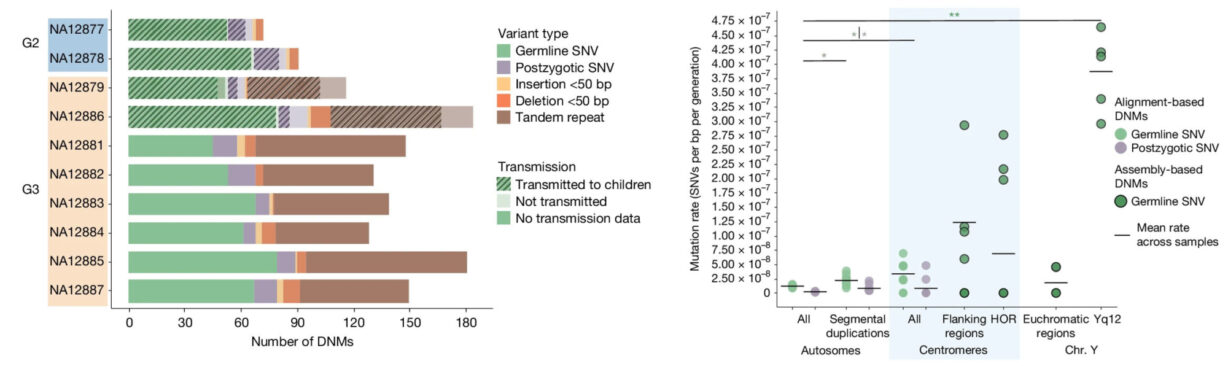
This multiplatform, multigeneration study revealed insights in the repetitive regions, uncovering between 98 and 206 DNMs per transmission, including single-nucleotide variants, non-tandem repeat indels, de novo indels, and centromeric DNMs.
The data revealed a strong paternal de novo bias, demonstrating between 70–80% of germline DNMs originating from fathers and reinforced a potential connection between paternal age and mutation burden.
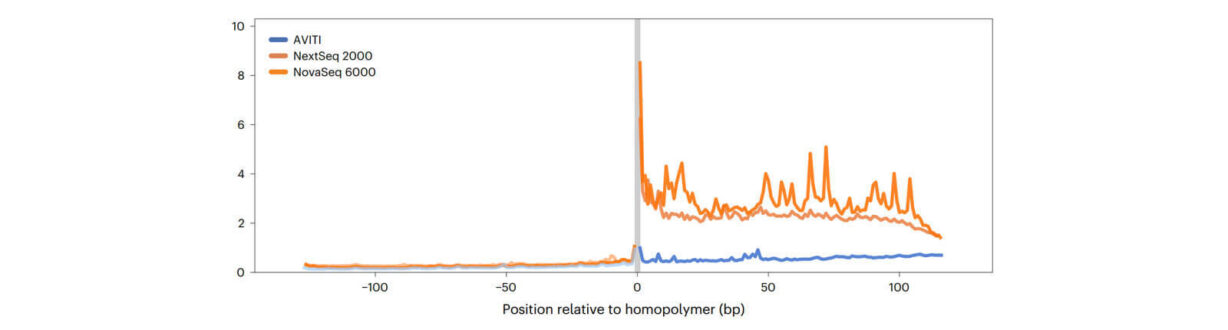
Interestingly, these mutation types didn’t distribute randomly. Repeats and complex regions that are historically hard to sequence show elevated mutation rates. That’s where high homopolymer accuracy and base-level fidelity really matter.
Summary of types (left) and location (right) of de novo germline mutations found in the CEPH 1463 study. Figure from Porubsky, D., Dashnow, H., Sasani, T.A. et al. Nature (2025). https://doi.org/10.1038/s41586-025-08922-2. CC-BY-4.0
ABC sequencing through homopolymers
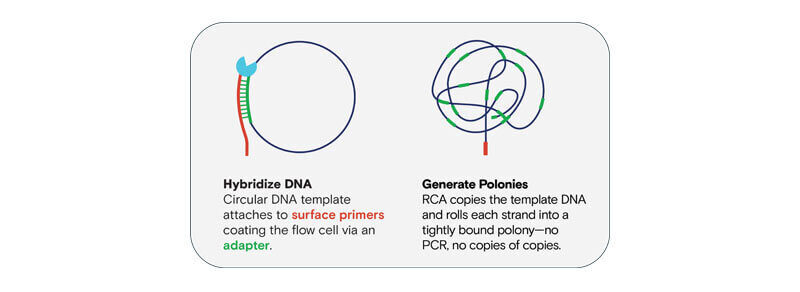
Many DNMs, like de novo tandem repeats, occur in homopolymer or other difficult to sequence regions. The AVITI system leverages our innovative avidite base chemistry (ABC) which uses rolling circle amplification to generate polonies from a single template, creating thousands of copies, so it can provide high-accuracy reads through homopolymers and repetitive regions—areas that are often skipped or miscalled by other technologies.
This study noted the low stutter in Element data at homopolymers and leveraged this high data accuracy to enable precise sequencing of traditionally challenging regions in the genome. By accurately reading homopolymeric areas, the AVITI platform enhanced the detection of mutations and structural variants that were once elusive. This advancement paves the way for deeper exploration of genetic diversity, particularly in regions critical for gene function and disease.
Rethinking inheritance maps
By placing these DNMs on a high-resolution recombination map, the authors tested whether de novo structural variants align with meiotic crossovers. The answer: not always. The expected correlation wasn’t present, suggesting that there is more to learn about the mechanics of recombination and structural change.
The study also found that 16% of de novo SNVs were postzygotic, pointing to early developmental mosaicism. These mutations can complicate interpretation in clinical genetic research and raise new questions around recurrence risk in families.
Looking Forward
The CEPH 1463 study highlights the diversity of human genetic variation and clarifies the mechanisms behind de novo mutations and structural variants. It challenges old assumptions, introduces new questions, and brings the field closer to understanding how genetic variation unfolds.
At Element, we’re proud to see AVITI powering the kinds of studies that make the future of genetics a little clearer. As we advance in our understanding of genetics, we continue to appreciate the complexity of our biology and what it means for health and evolution.
]]>urn:uuid:8f90df38-c398-4e86-9520-6607000cf644Uncovering Rare Variants and Spatial Heterogeneity: An Interview with Dr. Suzuki2025-06-04T11:18:00-07:002025-06-04T15:32:02-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
During the recent High Dimensional Biology Symposium in Taiwan, Dr. Yutaka Suzuki of the University of Tokyo delivered a featured talk on his latest research in cancer genomics. We took the opportunity to sit down with him for a separate, in-depth interview to learn more about how his experience using our high-precision sequencing and spatial technologies is accelerating his work. From identifying ultra-rare mutations to pioneering single-cell and spatial multiomics, Dr. Suzuki shared how the AVITI™ family—including the multiomics-capable AVITI24™—is helping to uncover new insights into cancer biology.
Detecting what others miss
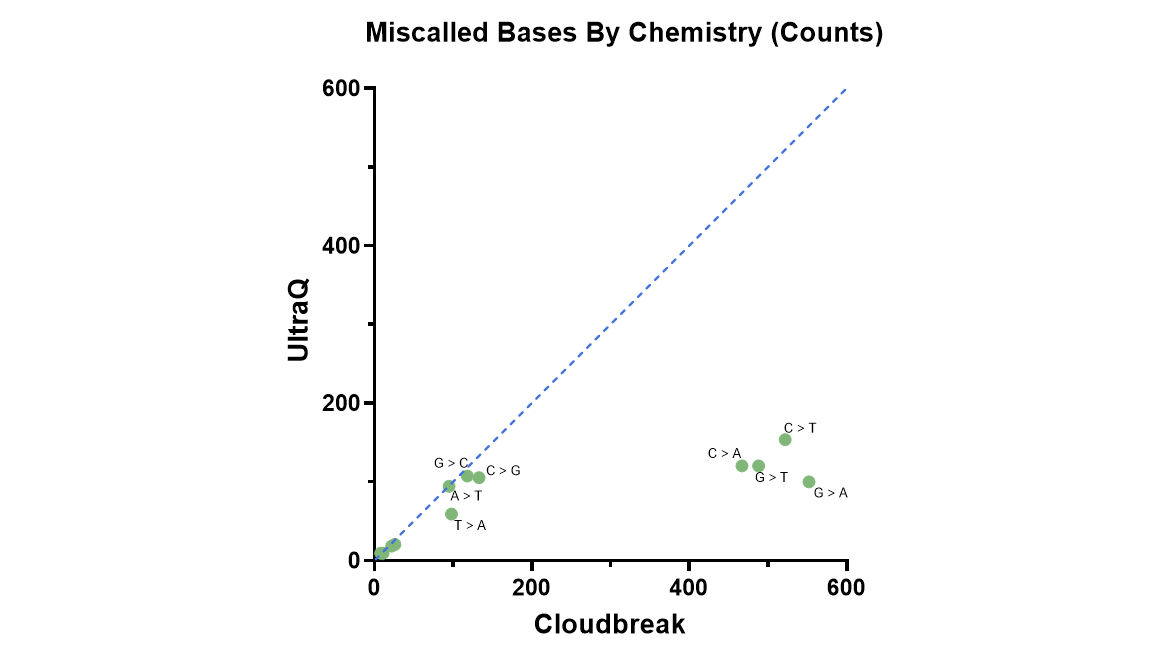
“When we first looked at the Q50 data,” Dr. Suzuki recalled, “I was really amazed.”
Using Cloudbreak UltraQ™ high-fidelity sequencing capabilities, his team was able to detect extremely rare mutations—those present in less than 0.1% of cellular populations. This level of sensitivity is essential in identifying minor cancerous mutations and conditions like clonal hematopoiesis of indeterminate potential (CHIP) in otherwise healthy individuals.
“Precise sequencing at this level wasn’t possible with other platforms,” he emphasized. “It’s indispensable for the kind of cancer research we’re doing.”
From hypothesis to mechanism: The power of AVITI24™
Dr. Suzuki, a pioneer of spatial analysis, described how his team has revealed remarkable diversity of gene expression in cancer samples—even within a single specimen. But understanding why those differences existed required more.
“That’s where AVITI24 came in,” he said. “Now we can perform drug perturbation and gene knockout analysis. Without that kind of validation analysis, we were generating hypotheses. With AVITI24, we can start uncovering mechanisms behind the expression diversity.”
Multiomics for a new era of cancer therapeutics
We asked Dr. Suzuki how spatial, single cell technologies were transforming the development of cancer therapeutics. He noted the need to think about cancer drug development from a new perspective. Currently, molecular-targeting drugs targeting driver mutations, like tyrosine kinase inhibitors, are reaching a plateau.
Spatial and single cell multiomics offer a new path forward. Multiomics analysis is a powerful tool to identify potential targets that control aberrant gene expression, “if we can find a way to rectify those aberrant transcriptome programs, there may be other ways to think about the treatment of cancer cells—beyond just targeting known mutations.”
Looking ahead, Dr. Suzuki is particularly excited about the new direct in sample sequencing capabilities on AVITI24, particularly the potential to reveal splicing patterns and immune cell interactions through TCR/BCR sequencing.
A collaborative partnership
Dr. Suzuki was candid about the initial skepticism he and his team felt. “We were concerned about the performance of Element because they were new to us,” he admitted.
But the transition was smooth, thanks to Element’s dedicated support. “They answered every question—no matter how basic—and even traveled from the U.S. to Japan for support and we successfully collected not only whole genome data, but also RNA, single cell, and ATAC sequencing.”
Dr. Suzuki concluded: “We couldn’t be more thankful for their dedicated support.”
A message to fellow researchers
“Don’t be too conservative,” Dr. Suzuki urged. “The sequencing platforms many of us are used to were designed for large-scale sequencing centers. But today’s research is more diverse and complex, and the applications are expanding widely. If we think about the multifaceted use of a sequencer for spatial analysis and high quality sequencing, the AVITI24 is a powerful solution.”
Dr. Suzuki’s journey illustrates how spatial, single-cell technologies and collaborative partnerships can push the boundaries of cancer research. From detecting rare mutations to decoding spatial complexity, platforms like AVITI24 are enabling scientists to move from observation to action—and from hypothesis to understanding.
]]>urn:uuid:d203eb74-af12-4e46-af6d-6105927e8fe6A New Study: High-Throughput Multiomics for the Modern High-Capacity Scientist2025-05-23T13:06:00-07:002025-06-11T11:02:44-07:00Michael Sullivanmichael.sullivan@elembio.com
At Element, our mission has always been to enable researchers to do “smarter science”. This means creating tools that optimize for time and resources, while maximizing the insights scientists obtain from their experiments.
Thats why we call AVITI24™ the first truly 5D multiomics system; a descriptor rooted in the system’s capacity to work with scientists to advance their research at the pace and scale modern innovation necessitates.
Traditional methods of omic analysis all suffer from the same common drawbacks: they only let scientists look at one aspect of cells at a time - either their shape, their RNA, or their proteins.
But AVITI24 changes this paradigm. As illustrated in this paper, the system enables scientists to analyze all of these aspects simultaneously in the same cells with single cell and spatial resolution, giving researchers a comprehensive view of cellular activity.
"It’s no longer enough to look at one data type in isolation," explains Tyler Lopez, lead scientist on the AVITI24 paper, and Element’s Associate Director of Biochemistry. "Without integrating protein, RNA, and cell morphology, critical insights are easily overlooked. You can have RNA responses without corresponding protein responses, or protein responses on a completely different timescale. The ability to move seamlessly between these data types is where the future of cellular analysis is heading, and what we are delivering with this system.”
In areas like cancer research, understanding exactly how cells respond to drugs is crucial. Cancer cells are incredibly adaptive, often finding ways to survive treatments that should kill them. This recent study demonstrates how AVITI24 reveals these survival mechanisms with unprecedented detail.
In our research, we exposed different cancer cell types to TNFα, a molecule that typically triggers cell death. Using AVITI24, we demonstrated that rather than failing to detect the signal, cancer cells actively rewire their internal signaling pathways to neutralize the threat.
"We made this system highly customizable from day one, a difficult but essential task. For example, our custom kit allows up to 88 antibodies. The sheer scale and flexibility we've created opens up entirely new possibilities for researchers across different fields."
For drug developers, this means faster, more accurate screening of potential treatments. For cancer researchers, it offers unprecedented insights into treatment resistance mechanisms.
By creating detailed maps of cellular responses over time, AVITI24 can help scientists identify precisely where drug interventions might be most effective. This could significantly accelerate the development of more targeted cancer therapies that outsmart resistance mechanisms.
Our scientists' next steps involve using machine learning to extract even deeper insights from these rich datasets, potentially revolutionizing how we approach drug discovery.
]]>urn:uuid:3c7c23c7-6ebe-4cf9-b1f4-960b70ac43d1Seeing deeper with optical pooled screening2025-05-19T10:33:00-07:002025-06-11T11:04:19-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
Biological research is evolving—combining targeted, hypothesis-driven studies with high-throughput discovery. By applying thousands of perturbations in parallel, pooled screening methods systematically uncover genes and pathways that drive key phenotypes such as survival, proliferation, or drug response. However, most pooled screens rely on sample enrichment based on a single endpoint measure through drug selection or fluorescence-based cell sorting and are limited to single-modality readouts, missing much of biology’s complexity.
Optical pooled screening (OPS)1 is a powerful solution to this challenge, bridging high-throughput perturbations with high-content imaging to unlock deeper biological insights. In this approach, researchers combine scalability with rich phenotypic information; however, its adoption is limited by complicated, manual workflows, limited plexity, and difficulty in incorporating additional readouts, like RNA and protein expression.
Here, we review the application and share how Direct In Sample Sequencing (DISS) on the AVITI24™ 5D multiomics system transforms OPS by creating a streamlined, integrated, and automated workflow with data-rich, multimodal results.
What is optical pooled screening?
Optical pooled screening integrates the scalability of pooled perturbation libraries with the rich, single cell phenotypic information typically obtained through microscopy.
In a typical optical pooled screen:
Cells are transduced with a library of genetic perturbations, each containing a unique barcode.
High-content imaging captures detailed morphology, protein localization, and cell-cell interactions.
The identity of the perturbation in each cell is determined through manual in situ sequencing of sgRNAs (<20 bp) by microscopy.
This workflow allows researchers to link complex cellular phenotypes directly to specific genetic perturbations at the single cell level in a massively parallelized manner. OPS presents many advantages over traditional pooled screens by providing rich morphology readout across millions of cells.
Challenges in current optical pooled screening workflows
While optical pooled screening offers many advantages, it introduces a range of workflow challenges that limit its widespread use. One of the primary hurdles is the complexity of the barcode readout. Accurate decoding of single cell perturbations requires labor-intensive, multi-step imaging protocols with intense manual processing. Without readily available commercial workflows, it relies on “homebrew” methods for cycling and in situ readout of the barcodes by microscopy.
Not only is this process time consuming, often taking 2+ weeks, it uses multiple reagent vendors, has limited z-dimension imaging, and only provides <20 bp of in situ sequencing. Because these methods rely on efficient hybridization to the barcode, there is often low detection rate of guides, often <40%.
The massive volume of high-resolution images generated during screens demands significant data storage, computational infrastructure, and sophisticated image analysis pipelines to extract meaningful information.
Together, these workflow challenges highlight the need for innovations that can streamline barcode detection and integrate data into a more seamless, high-fidelity pipeline.
A streamlined OPS workflow powered by AVITI24
DISS on the AVITI24 powers a fully automated, end-to-end workflow for generating OPS data. The pooled cell library can be plated directly on a Teton™ flow cell for fully onboard multiomic detection.
This approach integrates highly accurate in sample sequencing with multiplexed phenotyping capable of profiling cell morphology features, custom protein expression, and 3’ whole transcriptome (available in H2 2025).
See how we performed a 500 gene CRISPR screen, enabling large-scale, high-resolution profiling of perturbation effects and setting the stage for scalable, automated OPS studies. Download the poster.
Applications of optical pooled screening in biology and drug discovery
The versatility of optical pooled screening opens new frontiers across biological research and drug discovery:
Drug Discovery: Identify genes and pathways that influence subtle phenotypic traits and understand the mechanism of action or resistance of your research compounds.
Stem Cells and Development: Monitor shifts in differentiation status, uncover regulators of lineage specification, and map the trajectories of stem cells as they commit to specific fates—all by imaging key phenotypic markers.
Cancer biology: Changes in cell morphology, migration, invasion potential, or immune evasion can be linked to genetic perturbations. For example, uncover regulators of epithelial-mesenchymal transition (EMT) by screening for specific morphological signatures.
Neuroscience: Studying neurons and glial cells often hinges on fine-grained phenotypic features and OPS provides a scalable way to dissect the genetic regulators of these complex traits.
Functional Genomics and Systems Biology: Optical pooled screening allows researchers to build high-dimensional phenotypic maps that reveal functional relationships between genes, pathways, and phenotypes, accelerating the creation of comprehensive biological models.
In all these areas, optical pooled screening delivers insights that single readout pooled screens—or even single-gene imaging studies—would miss, opening new avenues for discovery.
The next frontier in functional genomics
As high-throughput screening continues to evolve, streamlined OPS workflows combined with AI-driven phenotyping will transform how we link genotypes to complex phenotypes.
By integrating imaging and transcriptomics at single-cell resolution, these multimodal screens offer unprecedented depth. Efforts toward overcoming current barriers and increasing democratization are making high-content pooled screens accessible to more labs, unlocking a new wave of discoveries across biology, drug discovery, and biotechnology.
References
Feldman D et al. Optical Pooled Screens in Human Cells. Cell. 2019 Oct 17;179(3):787-799.e17. doi: 10.1016/j.cell.2019.09.016. PMID: 31626775; PMCID: PMC6886477.
]]>urn:uuid:ea33e1d1-1276-45f8-9157-d07f2c20c193What’s next for AVITI24™: Continued innovation for your research2025-05-07T13:41:00-07:002025-06-11T11:16:14-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
Last year we introduced AVITI24, the first integrated platform for in situ multiomics and next generation sequencing. In 2025, we’re taking it even further with new capabilities, added compatibilities, and expanded flexibility.
We’re pushing the boundaries of what’s possible in spatial biology to unlock unprecedented multiomic insights—all in a single run, with no library prep, and ultra-fast results. While AVITI24 already offers robust capabilities, this year’s innovations elevate the platform to a new level of flexibility, scale, and scientific power.
Here’s what’s coming:
New Teton panels: deep phenotyping for immuno-oncology & neuroscience
For expanded use in neuroscience and immuno-oncology research, we introduced new pre-designed Teton™ panels. Each panel is designed to simultaneously analyze cell typing markers, cell signaling molecules, and disease-relevant drivers of cell function. With automated, onboard detection of 350 RNA transcripts, 50 proteins (including phospho-proteins), and 6 cell morphology markers, Teton fixed panels provide the reagents to advance your understanding of these important research areas.
Alongside the new fixed Teton panels, we’ve expanded sample compatibility to include cell suspensions, including T cells, B cells, and PBMCs.
With these optimized protocols and improved segmentation models, you can process hundreds of thousands to millions of cells in a single run, without compromising data quality from intracellular and surface markers. Our enhanced segmentation models even detect fine neuronal structures—like dendrites—that traditional models may miss.
Flexible protein customization
While fixed panels provide protein detection for 50 biologically relevant targets, we understand that you may need to study additional proteins for your research question. For this, we’re providing two paths to protein customization:
88-plex add-on panel Easily couple your own primary antibodies to Teton detection probes—without complex conjugation. Setup takes under two hours, and you can integrate custom probes directly into your fixed panel workflow for up to 138 protein targets in a single run.
24-plex mini panels For mix and match customization, we’re also providing pre-designed Teton mini panels containing 24 targets. Use our custom workflow designer and use up to 3 mini panels for the fastest route to protein customization.
Optimization kits: smarter starts, better data
To help you get it right the first time, we’re introducing a new optimization kit with our antibody screening kit so you can validate antibody performance before starting your run. Simply couple your antibody to Teton detection probes, add to cells, assemble into a Teton flow cell and image on your AVITI24.
For added use in drug discovery applications, our 48-well consumable supports high-throughput profiling of dozens of conditions—up to 96 samples in a dual sided run.
In one example, we profiled multiple cell lines across four treatments and ten time points. The result: detailed profiling of cell-type and state-specific responses—all in under a day.
Direct In Sample Sequencing: library-free, whole transcriptome + targeted RNA sequencing
On AVITI24, we’re excited to unveil a new era in in situ sequencing with Direct In Sample Sequencing (DISS)—no library prep required. Compatible with any species and any sample, we’ll offer fully automated RNA sequencing with reads up to 100 bp.
This groundbreaking tech includes:
3′ Whole transcriptome sequencing
Targeted RNA sequencing for hypothesis-driven panels
Compatible with simultaneous visualization of cell morphology and protein detection
First, we’ll enable targeted RNA sequencing for CRISPR guide or barcode sequencing and follow with the addition of whole transcriptome sequencing—all within a modular, customizable workflow.
Rapid multiomics customization
It’s not just about getting more data—it’s about getting the right data, faster and with more insight than ever before. With a fully modular cartridge and advanced software tools to design your experiment including a probe designer, you’ll have maximum flexibility to rapidly build fully customizable workflows with the read-outs and cycle length you need for your experiment. No need for time consuming custom solutions or white glove services, just flexible multiomics with a streamlined workflow.
Ready for your next breakthrough?
Whether you're decoding neurodegenerative disease, performing high-throughput CRISPR screens, or exploring novel immune states, continued innovations on AVITI24 give you the flexibility, scale, and depth to uncover what others miss.
The platform is evolving. Your science can too.
]]>urn:uuid:2659e36b-e19a-448e-ac15-b3ce66e61496Smarter Science, Faster Insights: AVITI24™ Takes Center Stage at AACR 20252025-05-05T11:54:00-07:002025-06-11T11:21:56-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
As the 2025 American Association for Cancer Research (AACR) annual meeting wraps up, we’re reflecting on the groundbreaking science, new collaborations, and the unveiling of what’s next in cancer research. This year, we were proud to showcase how AVITI24™, the world’s first intelligent multiomics platform, is transforming the pace and precision of cancer research and announce accelerated timelines for new Direct In Sample Sequencing (DISS) capabilities.
Throughout the conference, the energy at our booth was electric as researchers, collaborators, and partners gathered to see how AVITI24 is setting a new standard in multiomics with faster insights and smarter science.
A major milestone: accelerated rollout of Direct In Sample Sequencing
For us, one of the most exciting moments of the event was our announcement of the accelerated rollout of DISS, a transformative technology that enables spatially-resolved, single cell molecular insights directly in their biological context—all completely library-prep free. It unlocks research possibilities across new applications, from lineage tracing to CRISPR pooled screens and more. We can’t wait to see how cancer researchers will use it to advance their discovery.
Along with this announcement, we held daily presentations sharing all things AVITI24 and DISS in our meeting room. The talk, "Leveraging High Dimensional Multiomic Screens with AVITI24 to Accelerate Drug Discovery", highlighted ways researchers can now perform integrated multiomic analyses at scale, drastically shortening discovery timelines and expanding insights across biological systems. We took a deep dive into the DISS technology and shared real world data using the technology for optical pooled screens.
Innovation in action at our scientific posters
While new capabilities on the AVITI24 were a major focus at the show, our scientific team also shared their work to develop novel applications of our Trinity™ hybrid capture sequencing workflow for cancer research. If you missed our posters or want to take a closer look, we’ve made all the posters available for download.
Optical pooled screens with massively multiplexed phenotypic readouts on AVITI24
Optical pooled screening (OPS) is a powerful tool for linking morphological cellular phenotypes with in situ readouts of genetic perturbations. However, adoption of OPS has been restricted to a small number of labs due to the manual workflows and expertise required for data generation and computational analysis.
Here, we establish a fully automated end-to-end workflow for OPS on the AVITI24 system. We demonstrate highly accurate in sample sequencing of genetic perturbations combined with a highly multiplexed AVITI24 phenotyping panel to perform a 500-gene CRISPR screen for regulators of complex cellular processes. This work lays the foundation for automated OPS data generation, supporting rich profiling of perturbation effects at unprecedented scale.
Combined analyses of RNA transcript variations and comprehensive genomic sequencing allows for cross-validation of somatic mutations, improvement of sensitivity, and understanding of mutation effects on gene expression.
Here, we employed the novel Trinity target capture technology to simultaneously capture both RNA and DNA libraries in a single hybridization reaction. This optimized workflow significantly decreases sample-to-answer time and showed ~ 10% higher mean target coverage compared to data generated using conventional target capture workflow.
Mechanism of action discovery for cancer therapies with AVITI24
For decades, cancer therapy research relied on isolated analyses of transcriptomics, proteomics, or cell morphology to resolve heterogeneous responses. Integrating these readouts through multiplexed analysis enables a comprehensive understanding of how gene expression relates to functional outcomes and phenotypic effects, offering a holistic view for research of cellular processes impacted by treatment.
Here, we utilized AVITI24 to resolve complex systemic responses to tyrosine kinase inhibitors. By consolidating multiomic data in a single assay, this platform resolves multidimensional mechanisms underlying tumor responsiveness and drug resistance. By bridging molecular and morphology insights, our study provides a powerful framework for studying the intricate dynamics of targeted therapies, paving the way for innovative strategies to enhance cancer treatment and personalize patient care.
Hybrid capture technology is a powerful and cost-effective tool to detect somatic mutations in precision oncology research. Traditional hybrid capture workflows involve several time-consuming steps prior to sequencing which can delay results.
Here, we combine Trinity hybrid capture and rapid AVITI sequencing protocol to deliver targeted sequencing results within 12 hours. We applied this specialized Trinity workflow with a custom myeloid panel based on a design used by Genomics Medicine Sweden to detect mutations in reference DNA and patient blood, bone marrow, and fresh frozen tissue. This rapid, accurate, and robust solution for targeted sequencing offers significant advancements in turnaround time for hematology malignancy research.
To everyone who visited us at AACR 2025 — thank you. Whether you stopped by our booth, joined a session, or downloaded a poster, your passion for advancing cancer research fuels everything we do.
Let’s keep building momentum. Contact us to explore how AVITI24 can support your next breakthrough.
]]>urn:uuid:48f83e22-5d40-4216-9d59-074bf4c972b3Faster. Simpler. Smarter: How we designed a better hybrid capture workflow with Trinity™2025-05-01T11:02:00-07:002025-05-01T11:02:45-07:00Marina McCowin, PhDmarina.mccowin@elembio.com
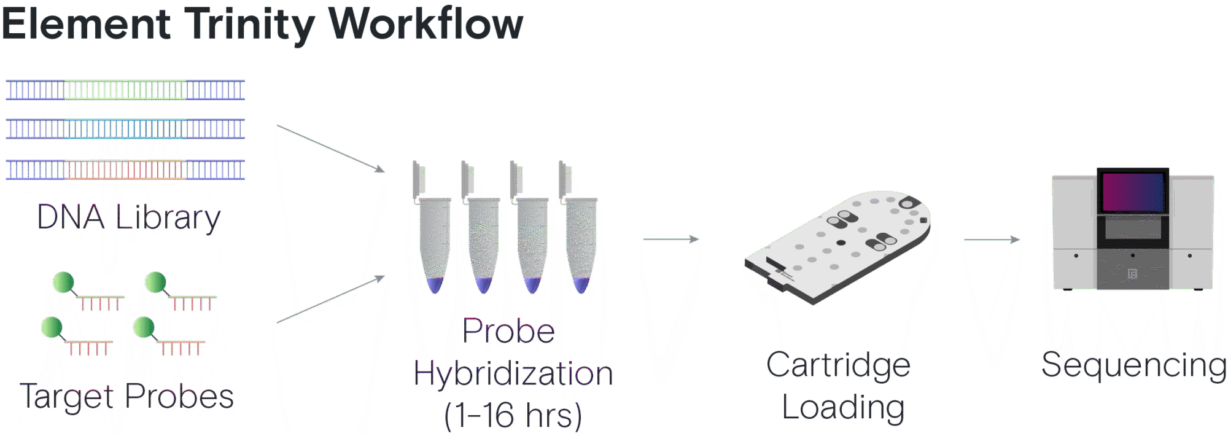
What is hybrid capture?
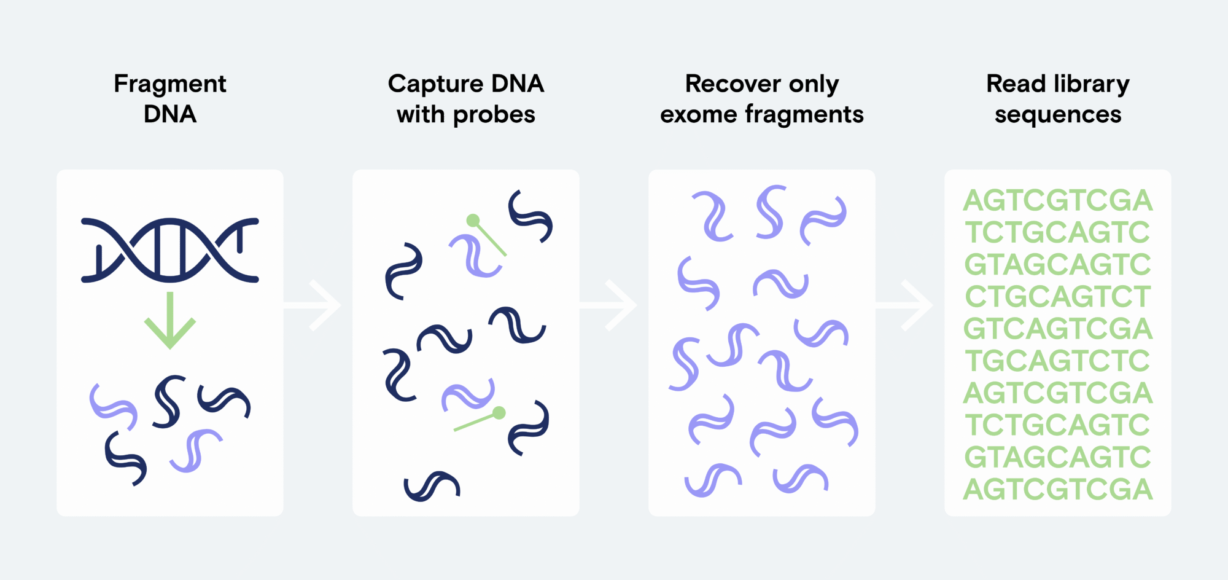
Hybrid capture is an essential biology technique used across many genomic applications, from cancer research and disease biology to microbial genomics, agriculture, and exome sequencing1-5. The hybrid capture process enables you to focus your sequencing efforts on specific regions of the genome, maximizing the sequencing reads applied to your regions of interest so you don’t waste reads.
Unlike whole genome sequencing, hybrid capture hybridizes your library to a set of probes that are specific to your application or organism. The probe set can be large or small, spanning just a few genes to an entire exome. This provides higher coverage for those regions of interest compared to a standard whole genome sequencing prep, allowing more samples to be sequenced on a single flow cell and reducing overall sequencing costs.
However, hybrid capture protocols can be lengthy and complex, and are often split into multiple workdays to complete2,5. After hybridization, traditional methods use magnetic beads and a series of temperature-controlled wash steps to isolate the hybridized material and remove non-specific material. While these steps help achieve a high on-target rate for your panel, they are also time-consuming and can lead to a large loss of DNA. Due to this loss, these workflows usually require PCR amplification just to generate enough material for sequencing. For some, the cost savings are worth the complicated manual procedures and extra hours in the lab.
In our recent pre-print publication about Trinity™, we show that you don’t have to compromise: the Trinity workflow on an AVITI™ system reduces the hands-on and total time required for a hybrid capture workflow, all while maintaining or exceeding the performance of traditional hybrid capture.
How is Trinity different?
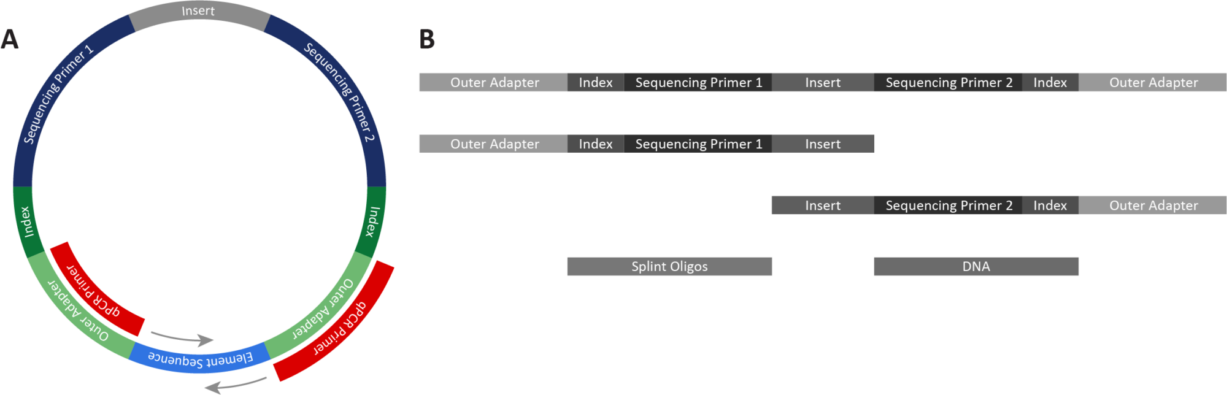
When the Element team first thought about how to improve hybrid capture workflows, we knew we wanted to decrease and simplify the manual steps in the process. The team developed a new flow cell surface and new reagents capable of capturing the library fragments bound to the probes, successfully eliminating all of the manual post-hybridization steps from the workflow.
Trinity still hybridizes the library to the same panel of probes as the traditional workflow, but the hybridized library can then be loaded directly onto the instrument. This means there is no bead capture, no time-consuming and finicky manual wash steps, and no post-capture PCR amplification needed!
The Trinity workflow reduces time required for hybrid capture workflows from multiple days to just 5 hours with a one hour fast hybridization.
Faster, easier prep with exceptional results
Eliminating manual post-hybridization steps provides a huge time savings in the hybrid capture workflow. While the traditional workflow had to be broken up into at least two days to complete all steps prior to sequencing run set up, the entire Trinity workflow, from library preparation through sequencing run prep, can be completed in as few as five hours using a 1-hour hybridization step.
To further validate the robustness of the technology, we tested multiple capture panels down to 700kb and found that Trinity’s performance was consistent for multiple vendors and across panel sizes. With the reduction in post-hybridization PCR and washing, we generally saw improved duplicate rates and library complexity, higher mean target coverage, and better variant calling benchmarking. We also present a unique opportunity to eliminate PCR entirely from the hybrid capture process and sequence a completely PCR-free exome with Trinity.
Choi, M. et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A 106, 19096-19101 (2009).
Gnirke, A. et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27, 182-189 (2009).
Kim, D.W., Nam, S.H., Kim, R.N., Choi, S.H. & Park, H.S. Whole human exome capture for high-throughput sequencing. Genome 53, 568-574 (2010).
Bansal, V., Tewhey, R., Leproust, E.M. & Schork, N.J. Efficient and cost effective population resequencing by pooling and in-solution hybridization. PLoS One 6, e18353 (2011).
Xiao, W. et al. Toward best practice in cancer mutation detection with whole-genome and whole-exome sequencing. Nat Biotechnol 39, 1141-1150 (2021).
]]>urn:uuid:8402b6aa-629e-48dd-9f7f-b79df0fccc135D Multiomic Insights with AVITI24™: A New Era of Discovery2025-04-21T12:15:00-07:002025-04-21T12:17:43-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
The world of multiomics is evolving. Traditional methods require multiple platforms, numerous samples, and extensive data integration to achieve a comprehensive view of biological processes. But what if there were a way to capture deep, multi-dimensional insights in a single experiment—without the need for complex workflows?
Enter AVITI24™, a breakthrough platform that enables high-dimensional co-detection of RNA, morphology, protein, and their dynamic response with spatial resolution. This platform is designed for discovery, empowering researchers to explore biology at unprecedented depths.
Gene expression analysis is central to multiomic research, and AVITI24™ enhances sensitivity and dynamic range. With automated onboard probe hybridization and amplification, the platform enables researchers to:
Profile 350 transcripts with high sensitivity
Detect thousands of transcripts per cell
This high-throughput capability accelerates transcriptomic research without the need for library prep or extensive sample preparation.
2. Protein Detection
Protein expression and phosphorylation play a central role in cellular function and signaling pathways. AVITI24 delivers unparalleled protein analysis by mapping:
Up to 138 cell surface, intracellular, and phospho proteins
Subcellular resolution using automated onboard antibody binding, amplification, and ABC sequencing
By providing this level of detail, AVITI24 helps researchers uncover complex protein interactions and regulatory mechanisms.
3. Morphology Analysis
Cellular structure plays a crucial role in function, development, and differentiation. With AVITI24, researchers can visualize 6 morphological features:
Nuclei, cell membrane, actin, endoplasmic reticulum, Golgi apparatus, and mitochondria
Multiplexed feature detection using reversible cell labels
Real-time segmentation and automated feature extraction
This integrated approach enables a more holistic understanding of cellular morphology alongside molecular insights, shedding light into the relationships between these readouts.
4. Dynamic response
Understanding how cells respond to signals, their environment, or compounds is critical in drug discovery, disease research, and functional genomics. In a single run on the AVITI24, you can:
Screen cellular responses over time
Analyze different compounds and dosages
Profile gene perturbations
With its scalable throughput, researchers can work with 1-well, 12-well, or 48-well formats (coming in 2025), allowing up to 96 samples to be profiled in less than 24 hours with a dual sided run.
5. Spatial Resolution
Knowing where biological molecules are within cells is just as important as knowing what they are. AVITI24 provides single-cell precision, spatially capturing analytes to reveal:
Intracellular locations of RNA and protein expression
The intricate organization of cellular structures
This spatial mapping capability enables a new level of insight into how biological components interact within their native environments.
The Future of Multiomics is Here
With AVITI24, we’re redefining what’s possible in multiomic research. By eliminating library preparation and integrating high-dimensional RNA, protein, morphology, and spatial data, researchers can now obtain a comprehensive, multiomic view of biology in a single experiment.
]]>urn:uuid:470b21b9-3a67-4f27-9659-d6949ca31cf1Women’s History Month Spotlight: Ruth Sager2025-03-25T08:47:00-07:002025-03-25T08:47:26-07:00Michael Sullivanmichael.sullivan@elembio.com
Here at Element, we recognize that science is not an automatic march forward, but rather the culmination of countless discoveries made across generations. The history of scientific progress is riddled with spectacular contributions from women scientists that far too often go overlooked or uncredited, yet have been essential to building our current understanding of human biology. This Women’s History Month is a reminder that scientific innovation is at its core a human endeavor, shaped by the unique perspectives of the men and women who pursue it.
A prime example of an often-uncredited female scientist is Dr. Ruth Sager (1918-1997), a pioneering American geneticist whose work challenged the dominant belief that genetic information resided only in the cell nucleus. In the 1950s and 1960s, while working with the green algae Chlamydomonas, Sager discovered and characterized what we call “non-Mendelian inheritance patterns”-- early evidence that DNA also resided in chloroplasts.
This research was groundbreaking, and established an entirely new field called cytoplasmic genetics. At a time when most scientists dismissed the possibility of genetic material outside the nucleus, Sager’s discovery was both bold and original. Despite facing significant skepticism from the scientific establishment, her persistence led to the acceptance of a fundamentally new understanding of genetic inheritance and evolution.
After establishing herself as a leader in non-nuclear genetics, Sager went on to reinvent her scientific career in her fifties, pivoting to cancer research at Harvard Medical School's Dana-Farber Cancer Institute. There, she made similarly significant contributions by identifying tumor suppressor genes, which can prevent cancer when functioning properly.
But as was far too common during this era, Sager was personally witness to significant gender barriers over her scientific career. Despite her immense contributions and discoveries, Sager did not receive a full faculty position until age 47.
Sager’s journey throughout her scientific career is an example of how intellectual courage and personal resilience can overcome stark obstacles. Sager twice established herself as a fearless pioneer in different fields. She changed our understanding of genetics while navigating the challenges faced by women in science during the mid-20th century.
At Element, we recognize the advancements of the AVITI and AVITI24 platforms would not be possible without the ingenuity and courage of scientists like Ruth Sager. Sager’s drive to prove the existence of DNA in chloroplasts not only expanded our understanding of genetics, it also dismantled 20th century preconceptions about who could make landmark discoveries in the first place.
So, on this Women's History Month, we celebrate Dr. Ruth Sager, whose persistence in the face of institutional resistance ultimately transformed our understanding of genetic material and paved the way for the multidimensional biological insights our platforms deliver today.
]]>urn:uuid:ed15b54e-70a7-4caf-9d83-ba943d23867924 Ways AVITI24™ High Dimensional Multiomics Can Transform Your Research2025-03-17T14:32:00-07:002025-04-25T10:46:38-07:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
Every layer of biology — from RNA to protein to cellular morphology — and their spatial and temporal relationships provides unique insights to give a more complete picture of cellular function, signaling, and disease states. However, understanding each of these layers through existing multiomics approaches often requires the use of multiple platforms, multiple samples, and the integration of disparate datasets. Our AVITI24 platform changes that by providing simultaneous multiomic readouts directly from your cells with next day results.
Having the right tools can make all the difference in your research. So, how can AVITI24 accelerate your discoveries? Here are 24 reasons why it’s the platform to take your research to the next level.
1. Dual capabilities: One instrument that does it all — in situ, high dimensional multiomics and next-generation sequencing.
2. Simultaneous high dimensional multiomics: With Teton™ CytoProfiling, you can detect transcripts, proteins (including phospho-proteins), and cell morphology features and understand their spatial and temporal dynamics.
3. High-quality NGS: Industry-leading accuracy and cost-efficiency with Cloudbreak™ sequencing, including flexible throughput options.
4. Innovative avidite base chemistry (ABC) technology: ABC technology was purpose-built for expanded flexibility and integrated multiomic readouts.
5. Visualize cellular morphology: Onboard, automated cell paint for 6 morphology features with reversible cell labels to enable multiplex feature detection.
7. Profile RNA transcripts: Subcellular spatial detection of 350+ transcripts with batched detection of 1000s of transcripts per cell.
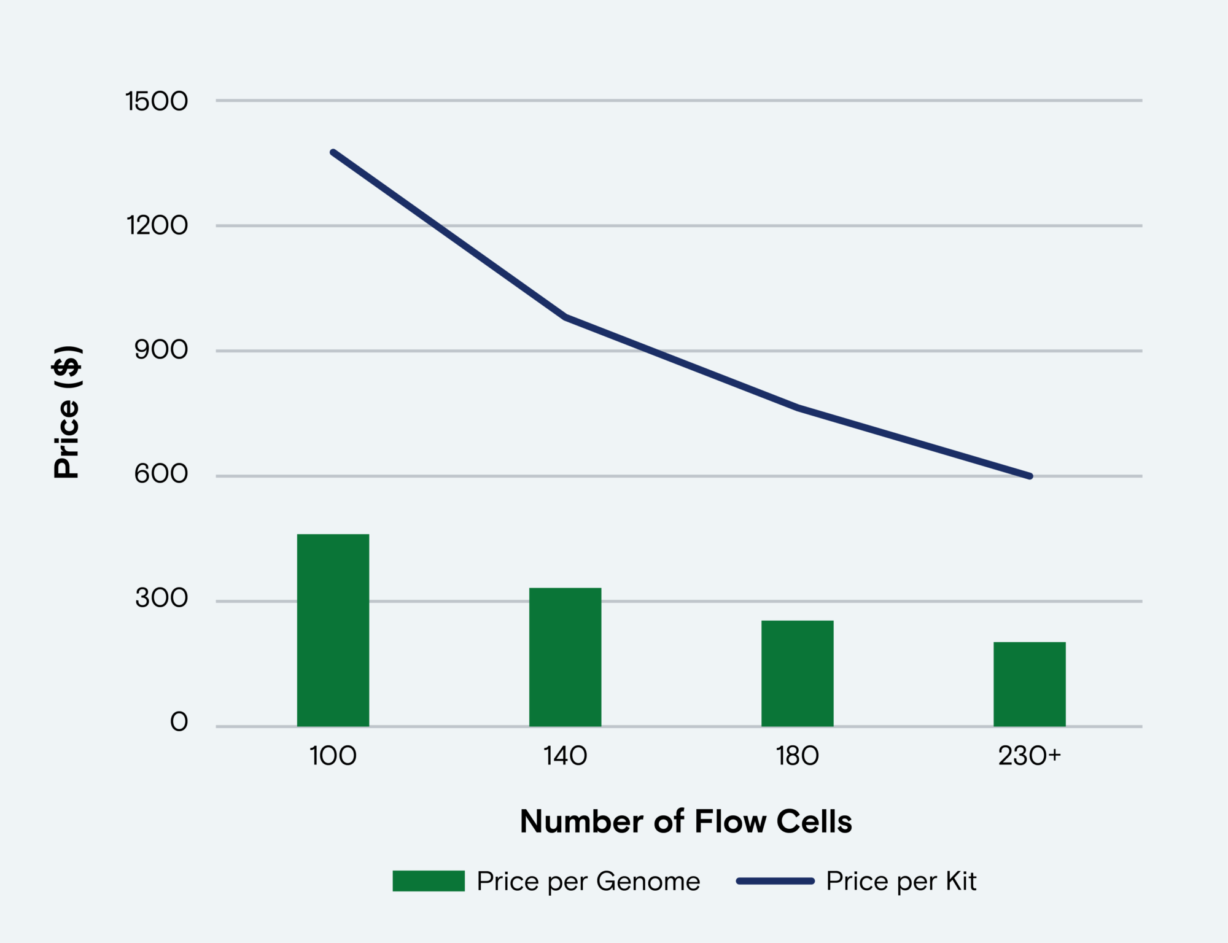
8. Increased sequencing power*: Achieve up to 3B reads per dual sided run with 50% greater sequencing output—at no additional kit cost or change to your workflows.
9. Library-prep free: Simply adhere your cells to the flow cell, fix them, and let the onboard detection chemistries do the rest.
10. Largest imageable area: With 20 cm2 imageable area in one dual-sided run, you can analyze more cells without the need for field-of-view selection.
11. Next-day results: Ultra-fast run times with less than 1 hour of off-instrument sample preparation and <24 hour instrument run times.
12. Automated, onboard detection: No need to incubate your sample with antibodies, probes, or other labels—protein, RNA, and cell morphology detection happens on board the instrument, so sample prep is less than an hour of hands-on time.
13. Industry-leading throughput: Process up to 2 million adherent cells per dual sided run and get full multiomic information about your cells in just one day.
14. Flexible sample inputs: Compatible with diverse sample types, including immortalized cell lines, primary cell lines, co-cultures, and cell suspensions.*
15. Multiple well configurations: Flexible flow cell configurations including 1 well, 12 well, or 48 well* so you can look at more samples, more timepoints, or more treatments in one experiment.
16. Curated pre-designed panels: Pre-designed panels for high-plex RNA and protein detection containing validated, biologically relevant targets to address your research questions.
17. Customization capabilities*: Build custom panels tailored to your specific experimental needs. Rapid protein customization requires no oligo conjugation to reduce complexity.
18. Sensitive and dynamic detection: Multiple rounds of sequencing enable detection of RNA and proteins with exceptional sensitivity—up to 1000s of counts per cell.
19. Subcellular, spatial resolution: Resolve thousands of RNA and protein counts per cell with spatial detail at sub-250 nm resolution.
21. Streamlined software tools: End-to-end software support, including cloud-based analysis, real-time monitoring, compatibility with community tools, and intuitive visualization tools for Teton™ assays.
22. Dual sided runs: Dual independent flow cells enable concurrent Teton CytoProfiling and sequencing runs with independent run starts for maximum flexibility.
23. Future-ready: AVITI24 is built to evolve with continued innovation, expanding capabilities and sample compatibility, without the need to replace the platform.
24. Comprehensive service and support: Backed by a dedicated service and support team ready to assist with fast turnaround times and scientific guidance.
AVITI24 brings together flexibility and speed to deliver unparalleled biological insights so you can get the most out of your samples and time in the lab. Whether you’re working with complex multiomic datasets, high-quality sequencing, or innovative solutions like direct, in sample sequencing, the AVITI24 platform unlocks more possibilities so you can focus more on discovery and less on experimental workflows.
*Expanded capabilities, enhanced sample type compatibility, and additional capabilities available on AVITI24™ in 2025.
]]>urn:uuid:c062e1a4-43f2-4fd2-b578-52aa20085be6Black History Month Spotlight: Dr. Ernest Everett Just2025-02-25T07:05:00-08:002025-02-25T07:05:31-08:00Michael Sullivanmichael.sullivan@elembio.com
Scientific advancement may appear on the surface like a certainty; an unstoppable force that only moves forward. But scientific innovation only happens due to years and years of compounding research contributed by individuals from all walks of life. People, and their curiosity, passion, and efforts are the center of our contemporary understanding of human biology.
During this Black History Month, we at Element believe it is important to recognize both the contributions Black scientists have made in the past, and the hurdles they have overcome during time periods where they saw great obstacles to their reputation and access to education.
One such scientist was Dr. Ernest Everett Just (1883-1941), an African American biologist and educator best known for his study of marine and cellular biology. Dr. Just’s schooling and educational career included stops at Dartmouth, where he graduated magna cum laude in biology; Howard University as a Professor in its Biology Department; and the University of Chicago, where he received his doctorate in zoology.
Dr. Just’s scientific legacy is rooted in his work with marine organisms, where he studied at length the movement of water into and out of living marine egg cells. His research proved vital in our understanding of internal cellular structures; a landmark finding that influences the modern development of life science technologies and tools. Dr. Just was the first to introduce the idea that cellular differentiation is not driven solely by the nucleus and genes present, but is also influenced by cytoplasmic factors.
Over his lifetime, Dr. Just published over fifty scientific papers and two influential books: Basic Methods for Experiments on Eggs of Marine Mammals (1922) and Biology of the Cell Surface (1939), which saw worldwide acclaim from the scientific community. His research has been foundational to the field cellular physiology, and many scientific advancements we benefit from today would not be possible without it.
At Element, we recognize our AVITI and AVITI24 platforms are built on the bedrock of centuries of scientific discovery and innovation. But scientific discovery does not happen in a vacuum. Scientists like Dr. Ernest Everett Just’s contributions in the field of cellular physiology went hand in hand with challenging the status quo of an era when racial barriers in America limited opportunities for Black scholars.
So, on this Black History Month, we raise a toast to Dr. Ernest Everett Just, who paved the way for a much deeper and richer understanding of our own biology.
]]>urn:uuid:f58812d5-dade-43a7-9486-742f8f9f4513Reflecting on JPM, and Element’s Road Ahead in 20252025-02-07T11:23:00-08:002025-02-07T11:23:58-08:00Molly He, PhDmarisabatey@gmail.com
The annual J.P. Morgan Healthcare Conference in San Francisco is in many ways our industry’s “Superbowl.” For us at Element, this past week’s conference was one of our most pivotal. We closed 2024 strong, over-delivering on our promise of doubling our revenue and AVITI installations compared to 2023.
On January 13th, we took the stage to highlight several new milestones and collaborations: Alongside Revvity, we will co-develop an in vitro diagnostics solution, focusing on newborn sequencing worldwide with our AVITI instrument. We also took another step into the proteomics market through a collaboration with Alamar Bio to offer a co-bundled proteomics solution. And we signed on as a commercial partner with the Human Cell Atlas consortium, an initiative working to map every cell type in the human body and expand our understanding of biology. These partnerships will prove instrumental in driving Element’s continued growth in the year to come.
But most importantly for Element in 2025 will be AVITI24: the world’s first fully integrated, library-prep free single-cell multiomics platform. It is a remarkable and revolutionary step for us, one that takes Element into a vast and untouched playing field. Through AVITI24, researchers will for the first time be able to generate multidimensional data from the same sample and same machine—completely transforming multiomics research and data generation, and definitively setting us apart from any other contemporary approach. Other solutions on the market are segmented and cumbersome, requiring post-experiment data integration. Not AVITI24. Our platform delivers higher data quality in a fraction of the time, all in one truly multiomic instrument.
AVITI24’s integrated platform, dramatically shortened run time, and capacity to co-detect multiple analytes —spanning DNA, RNA, proteins, protein modifications, and cell morphology— within the same biological samples is already carving out whole new markets for us. Since shipments began in December, I am pleased to share that 60% of installations come from new customers, including those in top-tier pharma. As AI is increasingly used to drive drug discovery, AVITI24 is uniquely positioned to provide the large-scale, unified data essential for researchers. By generating multidimensional data from a single sample at an affordable cost, AVITI24 is poised to overcome a major bottleneck in the field.
When we co-founded Element over seven years ago, we had a vision of a future where innovative scientific research tools would not be locked behind steep financial barriers. After all, how can we truly unlock and understand our biology if only a few can afford to access the tools required? So it is with great joy that I can say we are actually achieving this mission we set out on years ago through platforms like AVITI24.
JPM was about setting the stage. But there is more to come from Element in 2025. I hope you will join us on February 20th as we further unveil the road ahead for AVITI24 and our company.
]]>urn:uuid:d0b4d362-cb67-4721-816c-fbc93923d2ca5 reasons to watch Beyond and witness a new era of multiomics2024-11-21T11:24:00-08:002024-11-21T11:24:15-08:00Kelsey Swartz, PhDkelsey.swartz@elembio.com
Element held its inaugural virtual event, Beyond, diving deep into the science behind our newest technologies—our Trinity™ targeted sequencing workflow and the AVITI24™ next-gen multiomics platform. To kick off the event, our own DJ Freestyle entertained attendees and kept the energy high (check out his playlist!). From there, we took a closer look at the innovation behind our platforms, hearing directly from our senior staff and the scientists behind the technologies.
1. Discover the technology behind our AVITI™ family of systems and see how it set the stage for continuous innovation.
Our co-founders shared how we reimagined every element of a sequencing platform to deliver a solution designed to evolve with the researchers’ needs. From decreased run times to higher output to flexible read lengths to Q50+ quality—we’ve been able to integrate these solutions onto a single benchtop platform. Plus, they’ll describe how this set the stage for continuous innovation with simplified workflows, onboard library prep, multiomic cytoprofiling, and scalability.
2. Learn how Trinity is revolutionizing hybrid selection technologies with onboard library prep.
The team behind Trinity shared how we’ve modernized the targeted sequencing workflow by eliminating or moving all steps post-hybridization onto the AVITI platform. You simply load the post-hybridization material onto the sequencing flow cell and the AVITI takes care of the rest without any changes to the run time. With 1-hour fast hybridization options, you can generate next-day results with improved library complexity compared to traditional workflows.
3. Take a deep dive into the science behind the AVITI24.
We know that sequencing itself doesn’t provide all the insights into the complexity of biology. You typically have to perform several experiments to really understand RNA expression, protein expression and phosphorylation events, and show morphology changes in your cells. Our team describes how we combined these different aspects of biology in the AVITI24—the first platform that enables both NGS and in situ multiomics.
We share the workflow, describe how we benchmarked our data, and demonstrate how we sensitively detected a heterogenous drug response across cell populations by combining multiomic readouts. With just one hour of hands-on time, we studied over 1 million cells to gain new insights into the biology of drug response.
4. Hear from early access customers about their experience using these technologies.
You’ll hear why some of our existing customers chose an AVITI—from flexibility and compatibility with existing workflows to quality and performance to cost—the AVITI met and continues to exceed their expectations.
“They’re willing to take the leap into different advancements. That’s something that you only get from new key players that are not only changing the field, but they’re actually listening to what researchers what.”
-Hannah Dose, Co-founder & CEO, AU Genomics
Early access Trinity users shared how it transformed their current workflows. Chris Frazar, PhD, Norwest Genomics Center at the University of Washington, noted that their standard protocols for exome sequencing include an overnight hybridization followed by several rounds of stringency washing, but with the one-hour fast hybridization option of the Trinity workflow, they saved nearly a day and a half through the process.
Plus, you’ll see why William Lai, PhD, Assistant Research Professor, Cornell University, is excited about the unlimited potential of the AVITI24. Dr. Lai gave us a first look into his teams’ AVITI24 data examining DNA damage response across different cancer cell types. In a single experiment, the AVITI24 provided data regarding cell cycle status and detected proteins and transcripts that were not only common to all cell types, but also those unique to a specific cell type. As Dr. Lai noted, “it has the potential to be enormously transformative in terms of actually viewing and understanding everything that we’re doing.”
5. Get a sneak peek into the future of these technologies
The innovation doesn’t stop here. We also share what’s next for Element in 2025 — including direct in situ sequencing on the AVITI24. Our ABC chemistry enables us to move beyond counting RNA transcripts and unlock direct sequencing in cells. We share preliminary data of this technology including targeted sequencing of specific genes and an unbiased view of the transcriptome by sequencing the 3’ untranslated region. Importantly, we can combine direct sequencing with cell paint and protein detection for a rich multiomic view of cell biology.
Get all these insights, plus additional resources and infographics to learn more about ABC sequencing, the Trinity workflow, and the unlimited potential of the AVITI24 at our Beyond content hub.
]]>urn:uuid:a68e3c44-85ec-429f-95ff-2ab2c701183cNovel Insights into Single-Cell Transcriptomics with Paired-End Sequencing2024-08-06T07:00:00-07:002024-08-06T13:29:32-07:00Brian Coullahanbrian.coullahan@elembio.com
Single-cell RNA sequencing (scRNA-seq) is a transformative technology in biology that unlocks the complexity of cellular heterogeneity, developmental dynamics, and disease mechanisms with unprecedented resolution. Continued advancements in single-cell tools that better characterize gene expression from individual cells have further improved our ability to decipher these complexities, though technical limitations within the field of next-generation sequencing have restricted our ability to further explore this space.
Impacts of Homopolymer Regions on Sequencing Single-Cell Libraries
Challenges with standard SBS sequencing technologies have limited the use of paired-end sequencing of standard 3’ scRNA-seq libraries because of challenges associated with sequencing through homopolymers. Using 3’ priming methods for RNA-seq library generation, such as those provided by some 10x Genomics protocols, result in a library molecule where a portion of the poly-A tail is retained between the R1 sequencing primer and the coding region of the RNA. During standard clonal amplification on SBS sequencing platforms, these molecules undergo PCR using enzymes that introduce considerable error in these regions and result in clusters of molecules with a diverse length distribution of adenosine stretches. Sequencing through these stretches results in considerable phasing that can be seen in subsequent basecall results, yielding data of questionable utility. Because of this, standard sequencing scRNA-seq sequencing workflows rely solely on transcript data generated from the 5’ end of the library insert.
Exploring the Advantages of Avidite Base Chemistry (ABC) Sequencing in scRNA-seq
Element Biosciences has leveraged a different approach to amplifying library molecules on the surface of the flow cell, using rolling circle amplification (RCA) to generate polonies. As shown in the image below, a single molecule is used as a template by enabling the polymerase to extend the surface primer continuously through an isothermal process. By using a single template to create thousands of copies, error proliferation of difficult regions seen from PCR-based methods doesn’t occur, resulting in a highly uniform population of homopolymer length.
A team of researchers from the University of Utah School of Medicine noted the unique accuracy advantages enabled by ABC sequencing technology and evaluated the potential impact of paired-end sequencing to standard 3’ scRNAseq libraries in single-cell transcriptomics. The use of an AVITI™ system to overcome limitations of homopolymer regions was evaluated in a recent preprint posted to BioRxiv titled Improved characterization of single-cell RNA-seq libraries with paired-end avidite sequencing. The authors hypothesized that, by obtaining information from both sides of the RNA sequence, errors from misaligned reads and those that were unable to be uniquely mapped using single end data would be diminished during analysis. Additionally, by reading from the 3’ end of the transcript, polyadenylation site assignment could be more accurately defined and quantitated.
Increases in accuracy relative to SBS sequencing chemistry were well documented in the 2023 Nature Biotechnology publication Sequencing by avidite enables high accuracy with low reagent consumption, though this article highlights the first application of ABC towards true paired-end scRNAseq.
Post-homopolymer performance across platforms. Mismatch percentages of AVITI, NovaSeq 6000 and NextSeq 2000 reads before and after homopolymers of length 12 or greater.
Paired-End Sequencing of scRNA-seq Libraries
The authors generated 5 scRNAseq libraries using the 10x Genomics Chromium 3’ workflow and sequenced those libraries on both a NovaSeq6000 and an Element AVITI. Unlike standard 10x library sequencing schemes where 28bp are generated from read 1, allowing barcode and UMI identification only, the full 150bp read was generated from both platforms. As a secondary point of study, read 2, normally sequenced to 90bp, was also extended to 150bp.
Paired-end scRNA sequencing approach used by Chamberlin, et al.
Not surprisingly, results from read 1 generated on the NovaSeq experienced a significant drop in quality following the poly-A region where no measurable decrease in quality was observed with AVITI data. This further translated to an approximate 4x increase in unique alignments to the reference relative to NovaSeq.
Further analysis of paired end reads compared to standard single-end scRNAseq data, results indicated that, while the use of a second read did increase unique mapping statistics, the overall alignment rate was similar. The authors indicated the likely cause of the reduced impact of paired reads was short library length, though data did show a significant increase in base-pair precision of polyadenylation site assingement.
The Impact of Sequencing through Polyadenylation Sites
Several methods exist to predict polyadenylation sites based on RNAseq data, many are described in a 2023 publication, A Survey on Methods for Predicting Polyadenylation Sites from DNA Sequences, Bulk RNA-seq, and Single-cell RNA-seq. Advances in modeling, such as the use of deep learning algorithms, have improved the ability to do so, though all of these tools are based on the assumption that emperical sequence information from 3′ ends of mRNAs for direct profiling of poly-A sites remains a technical challenge. Although methods such as TAIL-Seq and PAT-Seq have been developed to do so, the workflows are considered to be extremely complicated and costly. To the contrary, paired-end sequencing of scRNAseq libraries using the AVITI enables a scalable, simple alternative to providing true emperical characterization of polyadenylation in parallel with single-cell gene expression analysis.
The ability to sequence through poly-A tails with high accuracy has implications beyond those noted by the authors. An area that may benefit from these advancements include the evolving field of mRNA vaccines, where accurate methods to QC plasmid templates containing the transcript sequence are required during the manufacturing process. Recent publications propose methods using NGS to do so, though a 2023 publication in Nature Communcations presented data highlightlighting challenges with SBS chemistry in accurately sequencing through the poly(A) region, a critical measurement required in mRNA vaccine QC.
Short-read sequencing correctly confirmed mRNA sequence, however, misalignment of short-reads at the poly(A) tail resulted in many errors and poor consensus accuracy and highlights the challenges of analyzing low complexity sequences with short-read sequencing. Gunter, H.M., Idrisoglu, S., Singh, S. et al. mRNA vaccine quality analysis using RNA sequencing. Nat Commun 14, 5663 (2023). https://doi.org/10.1038/s41467-023-41354-y. CC-BY-4.0.
Single-cell gene expression analysis has proven to be a breakthrough technique, ushering in a new era of discovery through profound insights into cellular heterogeneity, developmental processes, disease mechanisms, and therapeutic interventions. The ability to measure additional regulatory mechanisms of transcription like alternative polyadenylation has the potential of providing further understanding of cell-to-cell variability at the RNA level, though emerging single-cell multiomic approaches can further expand our view of systems biology beyond gene expression. Cytoprofiling capabilities of the Element Biosciences AVITI24™ platform have the ability of detecting and localizing RNA and proteins within cells and combine intra and extracellular morphology phenotypes to enable a more comprehensive view of individual cells at an unprecedented level. With the introduction of advanced tools like AVITI24, researchers can now map complex cellular pathways at a resolution that has not been previously obtainable.
]]>urn:uuid:2750aa29-98cd-40e9-ae9d-31024431c45eQ50+: Setting a New Standard for Sequencing Accuracy with Cloudbreak UltraQ2024-06-26T09:00:00-07:002024-08-07T10:52:44-07:00Semyon Kruglyak, PhDsemyon.kruglyak@elembio.com
A common measure of accuracy reported by sequencing platform providers is the fraction of data that exceeds an accuracy of 99.9% (commonly referred to as %Q30). The Element AVITI™ system leverages avidite base chemistry sequencing (ABC) to achieve a high fraction of Q40 data (99.99% accuracy) when applied to PCR-free libraries.1 However, certain applications may benefit from ever higher accuracy, in particular identification of low frequency variant alleles or reference genome refinement.
Setting a new standard, we developed Cloudbreak UltraQ™ with the highest accuracy specification on the market today. With improvements to the sequencing chemistry, a dramatic reduction of library preparation errors, and careful handling of analysis artifacts, UltraQ delivers 70% of reads at or above Q50 (99.999% accuracy) and 90% reads at or above Q40. As the measurement of very low levels of sequencing error is easily impacted by analysis methodology, here we take a deep dive into both our approach to reducing error and how we measured our progress.
How are Quality Scores Calculated?
Quality scores are assigned to each basecall during sequencing by mapping several predictors (such as intensity and phasing rate) to the quality score.2 The mapping is established through a machine learning model applied to multiple sequencing runs of samples from a well-characterized genome. The model inputs consist of the quality predictors for each base and a value of 0 or 1 depending on whether the basecall is an error or not. Any mismatches to the aligned reference sequence are considered errors. Given the predictor values and the correctness of each base, the machine learning algorithm generates the mapping from predictors to quality scores. That mapping is used to assign predicted qualities for all subsequent sequencing runs. Table 1 shows the relationship between quality score and error rate.
Quality Score
Error rate
Q10
1 in 10 bp
Q20
1 in 100 bp
Q30
1 in 1,000 bp
Q40
1 in 10,000 bp
Q50
1 in 100,000 bp
Table 1: Defining quality scores as a function of error rate p:
Verifying Quality Scores
However, the assignment of high quality scores to basecalls by a platform does not guarantee those quality scores reflect true accuracy. Quality scores need to be verified by comparing the predicted quality scores to empirically observed accuracy. This can be done using base call quality recalibration (BQSR)3, which is available as part of the Genome Analysis Toolkit (GATK)4 from the Broad Institute. An initial step in BQSR is to consider all bases assigned to a particular quality score, e.g., Q40, and then use alignment to determine the empirical error rate in that population. If the empirical error rate is 1 error per 10,000 bases, then Q40 accurately describes the population. If the error rate is more than 1 in 10,000 bases, the quality score is adjusted up or down to match what was empirically observed. This empirical quality score can be checked against the predicted score. If the predicted scores are accurate, then the plot of predicted vs. recalibrated quality scores is a straight line on the diagonal. Overprediction or underprediction would be characterized as points under or over the diagonal, respectively.[1]
Tracking Down the Sources of Error with E. coli
To attain Q50, we began by making improvements to the sequencing process. The improvements were focused on optimization of the sequencing recipe with respect to reagent concentrations, enzyme contact times, and formulations. These efforts led to higher average quality but did not increase the quality score to our target of Q50 in our benchmarking system. Where was the residual error coming from?
Careful characterization of error modes led us to library prep as the largest remaining barrier to significantly moving the needle on accuracy.5 Library preparation errors happen upstream of sequencing, so they are invisible to quality predictors and have a disproportionate impact on the recalibration of high-quality score assignments. The highest contributor to library prep-induced error was deamination damage, driven by library preps steps involving high heat. When a mutated template is sequenced accurately, a deamination event appears as a C to T base calling error. To mitigate these errors, we used a deamination reagent on the sequencer prior to polony generation. Library fragments with deamination damage (i.e., uracil bases) are digested and generate no sequencing data, eliminating a major source of confounding reads from downstream analysis.
The next highest error mode pertaining to library preparation is driven by end repair. End repair leads to template changes that predominantly show up in the first 15 cycles of R2. To address this, we implemented dark cycling at the beginning of R2, followed by 150 standard cycles. Since DNA fragmentation ahead of library prep is random, the skipped bases in one template are adequately sequenced in other polonies where they are positioned in regions of the template less likely to be compromised.
Figure 1 shows the impact of the deamination recovery agent and R2 dark cycling on errors with high quality scores in an E. coli assay that minimizes analysis-related errors. The baseline Cloudbreak chemistry data on the x-axis shows elevated errors in miscalls associated with deamination and erroneous end repair, whereas with UltraQ, on the y-axis, these error types are greatly reduced. With these changes, most bases in the model system exceeded Q50. In Figure 2, we see that the predicted Q scores tightly match recalibrated Q scores in both reads 1 and 2.
Figure 1: Normalized error count in E. coli comparing our baseline Cloudbreak chemistry with UltraQ. The deamination reagent mitigates the deamination errors (C to T and G to A on the reverse strand) while the dark cycling in R2 mitigates the end repair errors.
Figure 2: Predicted vs recalibrated quality scores for Read 1 (left) and Read 2 (right) in E. coli.
From Test Case to Use Case: Human Genome Sequencing
Demonstrating 70% Q50 with E. coli was an important milestone in our development, but bacterial genomes do not have the full contextual complexity of the human genome. However, extending the results to the far more complex human genome adds managing an additional error source: analysis artifacts. Given that errors are determined from alignment, an incorrect alignment can cause BQSR to treat accurately called bases as errors, thus reducing quality scores in the recalibration.
Incorrect alignments can occur due to mis-mapping of short reads to repetitive regions of a genome or because the reference itself is imperfect. Such issues are much easier to mitigate in E. coli where the entire reference genome is well established, and the repeat structure is known and relatively simple. To overcome analysis artifacts when benchmarking against the human genome, we turned to the CHM13 cell line. This cell line provides the full contextual complexity of the human genome but reduces analysis artifacts for two primary reasons. First, the cell line was used in the construction of the highly accurate T2T reference genome, eliminating most assembly errors.6 Second, it is a homozygous cell line, which obviates the need to mask heterozygous variants that could be confused for sequencing errors.
We sequenced the CHM13 cell line with our UltraQ reagents and, for the first time, observed over 70% predicted and over 70% recalibrated Q50 data in human (Figure 3). Table 2 illustrates the percentage of quality scores exceeding Q30, Q40, and Q50. We used third party tools for the recalibration and applied no BED files masking difficult regions of the genome. Instructions for reproducing our results are provided at the end of the blog.
Figure 3: Predicted vs recalibrated quality scores for Read 1 (left) and Read 2 (right) in CHM13.
Data exceeding Q threshold
Predicted
Recalibrated
%Q30
97.3%
96.8%
%Q40
92.9%
91.2%
%Q50
74.7%
72.8%
Table 2: Percentage of the data exceeding various Q score thresholds. R1 and R2 are averaged.
The development effort to attain over 70% Q50 data as benchmarked against the T2T human reference involved improvements to both library preparation and sequencing. To our knowledge, this is the first time that this has been achieved by any sequencing platform. By using multiple strategies to reduce the most abundant error types, UltraQ provides the firmest possible foundation for the development of highly sensitive assays for human health research.
Furthermore, the Q50 specification was met in both predicted and recalibrated quality scores, establishing a new standard for data quality. With the highest accuracy specification on the market today, UltraQ provides additional flexibility for AVITI users in applications where exceptionally high accuracy may be impactful.
Reproducing our Results
If you would like to reproduce our results or explore the data for CHM13, you can download the FASTQ files. We provide the entire FASTQ as well as a down-sampled version for faster compute. We align with BWA-mem with default parameters to the T2T reference found here: https://github.com/marbl/CHM13. The BQSR command that we use is below:
We compute recalibrated values using a “known sites” file that is blank (except for headers) but required by BQSR. We include it along with the raw data for ease of replication of our results.
What are the CHM13 cell line and the T2T reference?
The CHM13 cell line is derived from a complete hydatidiform mole (CHM), a rare event post fertilization where maternal DNA is lost and the paternal DNA is duplicated in its place, resulting in nearly uniform homozygosity across the entire genome. The simplified genome structure made it a natural choice for the Telomere to Telomere (T2T) Consortium to use for reference refinement. The T2T consortium used multiple sequencing technologies to generate gapless assemblies for all chromosomes (except Y) and correct error in the prior reference. Given the exceptional quality of the reference and the availability of the DNA that was used to generate the reference, sequencing CHM13 offers an ideal path to evaluating data quality in human, while avoiding nearly all analysis artifacts.
What is dark cycling?
Dark cycling refers to the ability to move along the DNA template strand without identifying the underlying bases. Because ABC separates the chemistry steps of base identification (avidity) and movement along the template (stepping), it is possible to omit avidity and imaging in specified cycles. Dark cycling iterates the incorporation of unlabeled, terminated nucleotides with removal of the block. Dark cycles take half the time of standard cycles, do not expose the DNA to additional rounds of laser light, and enable the user to skip over unnecessary or problematic regions of the template.
Arslan S, Garcia FJ, Guo M, et al. Sequencing by Avidity Enables High Accuracy with Low Reagent Consumption. Nat Biotechnol
42, 132–138 (2024). https://www.nature.com/article...
Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8(3):186-194.
Van Der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ Data to High‐Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr Protoc Bioinforma. 2013;43(1). doi:10.1002/0471250953.bi1110s43
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-1303. doi:10.1101/gr.107524.110
Kruglyak S, Altomare A, Ambroso M, et al. Characterizing and addressing error modes to improve sequencing accuracy. Published online February 5, 2024. doi:10.1101/2024.02.01.578321
Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. Science. 2022;376(6588):44-53. doi:10.1126/science.abj6987
[1]A further refinement in BQSR is to consider a set of covariates (e.g., position in the read, sequence context of the base) as well as the mismatch information as input into a logistic regression to refine the quality score values.
]]>urn:uuid:249faaa9-b5a4-4207-8062-b6688dddc4ebThe Potential of AVITI Sequencing in Genetic Research2024-06-17T09:00:00-07:002024-06-18T07:45:33-07:00Kristi Heimkristi.heim@elembio.com
While newborn genetic research holds immense promise for the future of healthcare, its success hinges on advancements in characterizing the genetics of rare diseases.
Today, about a quarter of babies born worldwide undergo some form of newborn sequencing, currently screening from a single to a maximum of 97 conditions for rare conditions. However, current tests cover only a fraction of the ~7,000 genes known to cause disease.
Revvity, Inc., based in Waltham, MA, develops these assays and methods and is at the forefront of utilizing AVITI sequencing in its research endeavors.
Newborn genetic research “delivers on the promise of population genomics, and our goal is to develop more assays to identify more disorders in newborns at an earlier stage," says Dr. Madhuri Hegde, Chief Scientific Officer at Revvity.
The Potential of AVITI Sequencing in Genetic Research
While still in its initial phase, complementing traditional biochemical and molecular assays with next-generation sequencing (NGS) into the research and development of newborn screening routines marks a significant advancement. NGS offers several advantages over traditional research methods including:
Multiplex testing: NGS allows for the simultaneous examination of multiple disorders, enhancing efficiency and coverage.
Identifying more genetic markers: researchers can better predict whether early intervention would benefit specific conditions.
Enhanced disease discovery: NGS can uncover diseases missed by previous methods, such as whole exome sequencing and curated panels.
For instance, research by Revvity published in JAMA Network Open and based on blood samples, found that genome sequencing identified a higher percentage of children at risk for pediatric onset diseases compared to exome-based panels.
This is where AVITI sequencing becomes a pivotal tool, holding great promise for advancing research that could lead to the development of more comprehensive genetic tests for newborns in the future. Revvity is now adopting AVITI in some of its labs around the world, including in the U.S., Sweden and India.
Implementing AVITI in Research Workflows
Revvity has developed an end-to-end workflow incorporating AVITI sequencing to bolster the research and discovery of previously unknown rare diseases. This workflow includes advanced dried blood spot collection and processing devices, nucleic acid extraction kits, liquid handlers, reagents for library preparation, and software for sample quality control.
Dr. Hegde explains the integration of AVITI sequencers: "A comprehensive cross-comparison showed AVITI’s throughput met our research needs perfectly, with improved chemistry reducing duplication rates. This efficiency means fewer reads are discarded, requiring less sequencing overall."
Impact on Genetic Research and Potential Therapies
As access to sequencing grows around the world, it can have a deep impact on global health.
One notable example of the benefit of NGS is in the research of Duchenne muscular dystrophy (DMD). The most common hereditary neuromuscular disease, DMD is caused
by mutation of the dystrophin gene, located on chromosome Xp21. Newborn screening for DMD involves an assay for the creatinine kinase enzyme, but research has shown that molecular confirmation is needed to determine if enzyme leakage is due to a defect in the DMD gene. This precise identification is crucial as pharmaceutical companies research potential treatments.
Conclusion
Revvity's use of AVITI exemplifies how cutting-edge sequencing technologies can propel our understanding and management of genetic disorders. As research continues to evolve, the integration of advanced sequencing systems like AVITI promises to unlock new insights, paving the way for more comprehensive and effective newborn screening protocols in the future.
“Newborn screening is probably one of the most successful public health initiatives,” said Dr. Hegde. “Discovering and developing new ways to test will only improve on that record.”
]]>urn:uuid:74b9b690-0f1b-4402-acd6-2caf1bc5faeaCornell Researcher, Epigenomics Core Director Wows NY “Shift Your Science” Roadshow2024-06-04T12:53:00-07:002024-07-15T13:03:33-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
While genome sequencing has given us a picture of the order of nucleotides—and the rearrangements that lie behind variation and disease—it has not provided a complete illustration of the complex interactions of genes, RNA, and proteins.
William Lai is working to change that, with help from Element’s AVITI™ system. An Assistant Research Professor in the Department of Molecular Biology and Genetics and Director of The Epigenomics Core Facility at Cornell University, Dr. Lai presented his research and his core activities at the Element Biosciences Road Show’s New York City stop.
He also shared why he was more than happy to switch from Illumina to AVITI sequencing.
Lai’s own research focuses on designing novel assays that will further our understanding of the fundamental mechanisms of gene regulation. “DNA sequence, local and distal chromatin (protein) and RNA all play both cooperative and antagonistic roles in when and how often any gene is transcribed. Understanding how these biological networks make decisions is the key bottleneck in understanding how perturbations to these systems result in a disease phenotype,” his Cornell research focus states.
Lai now uses chromatin immunoprecipitation (ChIP) to investigate protein-DNA interactions. This technique allows him and his colleagues to see where proteins bind to the genome, how they turn functions on and off, and how “gene regulation gets messed up.” Gene sequencing is critical to this research. Since about 2006, Lai said he has always had access to a gene sequencer. “If you want to make an assay, you need access to a sequencer,” he says.
The recent acquisition of an Element AVITI platform at Cornell has helped Lai accelerate his research. “With 24-hour turnaround around and better readouts, this allowed me to advance my research and develop new technologies.” Using AVITI will also help the core lab slash its sequencing costs from about $130,000 to $15,000 for the same number of reads.
Lai presented results his lab has produced recently, combining sequencing on the AVITI platform with improved bioinformatics tools and the appropriate biochemical techniques. The system is now part of a robotic platform in his lab and in the core lab he directs. Now, he can survey 96 target proteins on a single plate and generate 1.2 billion reads “in one go,” he said. This allows the simultaneous testing of multiple drugs, and of more factors that influence drug action, faster. “Because the AVITI is right here, I can get an answer in a week instead of a month.”
Pharmaceutical research, and cancer drug research in particular, has suffered from discovery that’s based more on brute force than a careful elucidation of functions. Lai has been looking at the perturbations caused by two anti-cancer drugs: flavopiridol, a CDK9 inhibitor which has been used in dozens of clinical trials (and failed most of them), and triptolide, a transcription initiation inhibitor which is now in a phase II clinical trial.
The two drug candidates stop RNA expression in cancer cells. But the way they stop transcription is very different. “You see a buildup of proteins,” Lai said. “The three-dimensional structure of DNA and protein changes depending on the drug perturbation. We want to see what is actually happening, because these actions have implications for how well the drug will work as a cancer therapeutic. Epigenomics of protein-protein, protein-DNA interactions…if we understood this, it would be easier to design a therapeutic at that point.”
We’re proud that AVITI sequencing is helping advance his knowledge (as well as that of his core lab customers) in this important area of research. “You’re either innovating or being left behind,” he added.
Why did the Cornell Epigenomics Core Facility leave Illumina?
Making a decision like purchasing a sequencer can be expensive and risky. For high capital expenditures, researchers understandably look for a trusted supplier, reliable operations and great customer support and service. Illumina looked like a logical choice for Cornell, until, according to William Lai, it didn’t.
Lai was looking to see what other sequencing technologies offered, and he came upon Element and AVITI. “One thing was shocking during the demonstration,” he said. “Our libraries broke the AVITI platform!” After a substantial amount of research, debugging and consultation with Element’s team, they found the problem—a synthesis flaw, specifically a truncation--in oligos they had ordered. The AVITI System and the Element support team found the synthesis error in a critical assay component, which the Illumina outsourcing provider had previously been sweeping the under the rug.
“We had expected 400 million reads but were only getting 200 million. We weren’t getting those all at once, but something seemed off,” Lai recalled. Illumina was filtering the errors, and not indicating them as such.
On top of that, runs on Illumina systems cost $7.85 per one million reads, while AVITI cost 90 cents per one million reads. Once the truncated oligo errors were resolved, the cost difference became even greater. Lai estimated that sequencing on Illumina cost $130,000 in 2023. AVITI costs for the same reads? Just $15,000.
]]>urn:uuid:6447df81-2977-41ab-a838-fc49b1335737AVITI24 — Six Ways to Shift Your Science to Cytoprofiling2024-05-15T14:31:00-07:002024-05-20T15:44:53-07:00Abbey Cutchinabbey.cutchin@elembio.com
The Element AVITI™ system redefined every aspect of sequencing to push the boundaries of performance, flexibility, and affordability. Now, AVITI24™ pushes benchtop sequencing into the next frontier by re-imagining core sequencing components, such as avidite base chemistry (ABC) and high-resolution imaging, to enable comprehensive multiomic analysis by Teton™ CytoProfiling, all on one platform. Teton CytoProfiling unlocks multiomic in situ sequencing in single cells, providing an unprecedented view of cell morphology and function.
Here are the six ways AVITI24 can Shift Your Science:
AVITI24 for core sequencing—now more powerful, with two billion reads across dual flow cells, more accurate, with up to Q50-level data quality with UltraQ™, and simple, with on-flow cell target enrichment and no library conversion step needed. AVITI sequencing brings the power (and affordability) of high-throughput systems straight to your benchtop, and industry-leading sequencing quality enables high accuracy variant calling, while reducing the data output required.
AVITI24 with Teton CytoProfiling — unite the diverse worlds of molecular biology and cell imaging. Now you can analyze DNA, RNA, proteins, phosphorylated proteins, and cell morphology simultaneously from just one sample in <24 hours. Teton is compatible with a wide range of sample inputs, including on-flow cell cultured cells and cell suspensions. With 10 cm2 of usable area per flow cell, Teton supports high-throughput, low-cost single cell experiments with up to one million cells per run.
Teton CytoProfiling: Cell Paint — cell morphology provides a unique view into cell phenotype and function, and changes in morphological profiles provide a powerful tool to study mechanisms of disease progression, drug response, and toxicity. With Cell Paint on Teton, profile up to 20 morphological markers for multi-feature cell segmentation and deep morphological analysis of cell types and states.
Teton CytoProfiling: RNA — profiling gene expression changes in single cells has emerged as a paradigm-shifting approach to resolve cell heterogeneity. Targeted RNA panels on Teton, focused on MAPK apoptosis and cell signaling pathways, immunology, and neuroscience applications, enable researchers to dissect cell types, states, and pathways at high resolution across tens of thousands to a million cells per sample. Enable wide-ranging applications from high-throughput drug screening and genetic perturbation studies to immune monitoring.
Teton CytoProfiling: Protein — protein is crucial to defining cell type and function. Targeted Teton protein panels enable profiling of up to 50 unphosphorylated and phosphorylated proteins within a single sample. Integrate morphology, gene expression, and protein expression for a multidimensional view of cell identity.
Teton CytoProfiling: In vitro ABC sequencing — profile up to 100 bp of in vitro sequencing directly in intact cells and unlock the ability to perform untargeted transcriptome analysis for unbiased discovery of cell types, states, and genome-wide transcriptional patterns. In vitro ABC sequencing also allows for profiling of genetic mutations at single cell resolution for deeper understanding of clonal evolution or therapy resistance.
AVITI24 brings you sequencing and cytoprofiling in a single integrated biology platform. With two independent sides of the instrument, it's now possible to perform sequencing and cytoprofiling experiments simultaneously. It is the only platform capable of on-flow cell culture, integrating rich molecular and cell imaging information into standard cell biology workflows. This integration allows for comprehensive multiomic analysis, connecting genotype, phenotype and function in each cell – that's true cytoprofiling.
]]>urn:uuid:c0cbce0c-98dc-4327-892b-6b4ba8848adbExploring the Human Body’s Own Drug Library2024-05-02T11:44:00-07:002024-05-02T15:15:56-07:00Kristi Heimkristi.heim@elembio.com
In the bustling labs of Infinimmune east of San Francisco, a revolutionary idea has taken shape: that within each human lies a treasure trove of potential antibody drugs waiting to be discovered. Every day, within our own bodies, nature orchestrates the equivalent of 100 billion antibody clinical trials, showcasing the ingenuity of our immune system’s response to potential threats. Infinimmune is harnessing this phenomenon in research to unlock and deploy protective antibodies as antibody therapeutics, with AVITI™ emerging as an indispensable tool in this quest.
“We started Infinimmune because we saw a huge opportunity to develop drugs that have already been validated for their safety and efficacy inside the human body,” said Wyatt McDonnell, a co-founder of Infinimmune and the company's CEO.
Humans have known about the power of the immune system since the time of the ancient Greeks, when Thucydides wrote about the plague in Athens. Since the discovery of antibodies over a century ago, antibody-based drugs have emerged as formidable weapons in the fight against diseases, notably cancer. Yet, despite this wealth of knowledge, the process of discovering effective therapeutic antibodies remains inefficient—and the drug discovery process contains many traps that can waylay a promising antibody drug candidate. Animal models—the workhorse of developing antibodies and other therapies—often do not provide a clear path to successful drug discovery, due to complex differences in how animals process proteins and other molecules.
In the current approach to antibody research and development, scientists test drugs that bind to a target without necessarily knowing if the target is a good one. Then, even if the target turns out to be relevant, the drugs themselves may not be effective in humans, even if they work in animal models of a given disease. Moreover, today’s technologies frequently produce antibody drugs for humans from transgenic animals or microbial systems.
Infinimmune wants to bring safer and more effective antibody medicines to the clinic by using completely human-derived antibodies, which would bring multiple advantages. These antibodies are less likely to trigger immune reactions when given to patients because the immune system already recognizes them as ‘self’. This reduces the risk of adverse reactions and improves safety and efficacy by minimizing damage to healthy cells and tissues—in part because these antibodies have already been “tested” inside a human body already. This approach also has potential to help discover novel, biologically important targets and improve matching of patient populations to immunotherapies.
“We believe that every company should be screening human antibodies to find new medicines for humans,” McDonnell.
Enter AVITI—a critical component of Infinimmune’s discovery research workflow.
“This is our bread and butter of how we find drug candidates every single day,” McDonnell said. “AVITI is, in one word, incredible. AVITI gives us NovaSeq-level pricing at NextSeq-level throughput, with an excellent yield of ultra-high-quality reads.”
For a company focused on sequencing only the cells that matter the most, affordable sequencing no longer must come at huge scale. The higher quality of avidite base chemistry is already helping Infinimmune researchers get farther with fewer reads.
“We are able to get more cells onto the instrument, we are able to recover more antibodies, and we are able to run our platform on larger numbers of samples,” McDonnell said. “That’s all hugely helpful in making our sequencing operations and our discovery campaigns more efficient.”
The team has already discovered new protein sequences in the Fc region (the antibody’s tail section that connects with cellular receptors and confers functional activity) that are relevant to engineering better antibody drugs, as the technology they’ve developed analyzes antibodies from the body directly rather than from a display library or a transgenic mouse.
“We found a massive amount of diversity in the Fc region. We are sitting on thousands of new coding, protein sequences of Fc. Wow!” McDonnell said. “We can observe this variation even with relatively shallow sequencing of a small number of humans. And sequencing the antibody regions with AVITI helped us identify this diversity.”
Infinimmune announced a partnership with Grid Therapeutics earlier this year to identify new drug candidates for non-small cell lung cancer using Infinimmune's Anthrobody™ drug-discovery platform and its Complete Human™ immunosequencing technology.
What inspires the team at Infinimmune is a combination of personal experiences and a profound sense of purpose. McDonnell and his fellow antibody drug hunters met while working at 10x Genomics. Having witnessed the toll of aggressive pediatric lymphoma, Huntington’s disease, rheumatoid arthritis, and chronic viral infections within his own family, McDonnell was driven by a determination to alleviate suffering and pave the way for a brighter future. The team also saw a clear opportunity to apply high-quality technology that traditional pharma and biotech companies have been slow to adopt.
“The reason people don’t develop autoimmune disease and cancer more often,” he said, “is that our immune systems do a wonderful job keeping these things at bay for decades at a time. And we need to learn what antibodies are responsible and then bring them to others.”
“Over time, we should be able to read out patients' immune systems as part of how you match the patient to the immunotherapy they receive, while ensuring that they receive extremely safe and extremely potent antibody therapy. That is the long-term future we would love to see.”
]]>urn:uuid:2ac0016e-66df-4941-a21c-ab45454de3b2AVITI™ for Agrigenomics Grant Winner to Study Plant Adaptation to Climate Change, Environmental Stresses2024-04-09T04:00:00-07:002024-04-09T10:56:58-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
How do plants adapt to stresses such as climate change and habitat alterations? What genes help plants make these crucial adjustments? Today, these questions have become more important than ever as the combined challenges of population growth and climate change make it urgent to secure the global food supply.
The AVITI for Agrigenomics Grant, first announced in January, presents an opportunity to push genotyping farther and faster with free sequencing services from Element Biosciences and Daicel Arbor Biosciences. For the 2024 AVITI for Agrigenomics Grant, we chose Dr. Jacob Landis, research associate at Cornell University’s School of Integrative Plant Science.
Genetic adaptations to environmental change
Landis’ winning proposal examines the genomes of the Andean blueberry—a wild plant native to the Andes Mountains in Ecuador—to see how they change in reaction to changes in altitude and climate. The blueberry (also called mortiño) lives between 5,000 and 14,800 feet above sea level and is an important part of the Andean diet and culture. It contains a wealth of anthocyanins, antioxidants and flavonoids and is used widely in traditional drinks, ice creams, wines and preserves. It also has been used in some medicines. The plant is well known for its ability to recover from deforestation and fires in high altitude ecosystems.
Landis, in collaboration with researchers at University of San Francisco in Quito, Ecuador, will use the AVITI for Agrigenomics Grant award to sequence four populations of blueberry, two at low elevation and two at high elevation. In each population, the researchers will sample 10 individual plants. The grant, offered jointly by Element Biosciences and Daicel-Arbor Biosciences, awards Landis and his colleagues two flow cells of sequencing and up to 48 library preps. Once sequenced, the researchers will correlate any differences with plant phenotype and environmental data. “The Andean blueberry can adapt to rapidly changing environments,” Landis said. “What areas of the genome are under selection? Why are the samples at higher altitudes successful up there?”
From mountain sample gathering to sequencing
Currently, the Ecuadoran researchers are gathering samples in the Andes. Previous research has shown that berry plants growing successfully at higher than 13,000 feet form a unique genetic cluster, regardless of whether they originated from lower or higher latitudes. They also noticed that at higher altitudes, genomic heterozygosity decreased.
For Landis, who had not studied the Andean blueberry before, the grant opens the door to unusual experimentation. “The plant lives at different elevations, and we want to see how the genome is responding to climactic changes. It’s a nice experimental setup without setting up the experiment, because the setup is already there!” he said.
Once the Ecuadoran team has completed sampling, they will send the DNA to Daicel Arbor Biosciences for sequencing on their AVITI system. They hope to have DNA ready in a month or so and are aiming at presenting their findings next January at the Plant and Animal Genome XXVI Conference (PAG). The Element/Daicel grant is also making sequencing in developing countries more accessible, Landis said. “Sequencing is a lot more expensive there,” Landis said. “We looked to AVITI as a less prohibitive way to sequence these samples.”
We are delighted to support Jacob’s work in genomics research and in helping introduce advanced sequencing techniques to countries like Ecuador. This work has the potential to help agricultural researchers and crop breeders better understand how plants and animals react to climate changes.
]]>urn:uuid:80811a51-4555-458b-9e00-62f0924d54f7Embracing the Library Prep Paradox2024-03-27T07:00:00-07:002024-10-31T08:01:42-07:00Shawn Levy, PhDshawn.levy@elembio.com
While the last 15 years have seen only modest increases in the diversity of sequencing instruments, the variety of applications and library prep methods has exploded. All the options for preparing samples and analyzing data create a challenge: how to embrace a promising new instrument without missing out on the wealth of applications?
Fortunately, from niche applications to whole-genome sequencing (WGS), library prep is premised on basic principles. DNA or RNA samples are fragmented into short, near-uniform segments. Then, additional sequences are added to identify the samples and enable amplification and base detection. Through this lens, library prep is not a challenge, but an opportunity. Element Biosciences leverages these principles to enable the full diversity of library prep without disrupting your workflow.
Entry points to ABC sequencing
Our avidite base chemistry (ABC) employs a unique amplification method, rolling circle amplification (RCA), that supports any library as a template for sequencing. Swapping PCR-based amplification for RCA not only improves quality, it establishes ABC as the first and only sequencing technology to encompass all next-generation sequencing (NGS) applications and partners across the field of genomics. The entry points to ABC are both broad and simple: adapted or native library prep.
Adapt with Adept™
Adapted library prep takes an already-prepared, third-party library and follows a short protocol to structure the library into an ABC-friendly format via circularization or amplification. The complexity and proportions of the library remain largely unchanged.
The Element Adept Library Compatibility Workflow is simple and lets you capitalize on ABC without giving up your preferred library prep and analysis. One noteworthy benefit of our continuous innovation is the 50% reduction in the amount of input library that the Adept Workflow requires. This low input enables most third-party individual or pooled libraries, both of which are Adept-compatible.
Dozens of third-party library prep and index kits have demonstrated compatibility with the Adept Workflow. The few incompatible libraries are easily made compatible with a few cycles of PCR. Whatever your prep, Adept makes it work with ABC.
Evolve with Elevate™
Native library prep generates libraries from nucleic acid input. This more familiar method is achieved with the Element Elevate Library Prep Workflow, which offers a growing range of library prep and index kits. LoopSeq™ for AVITI™ offers 16S and amplicon library preps, merging LoopSeq and Elevate chemistries to bring long-read capabilities to the AVITI System.
Founded on the core principles that have fueled 15 years of advancement, the Elevate Workflow departs in one meaningful way: it provides Elevate indexes and adapters. Pair these Element sequences with your Elevate library prep reagents to go end-to-end with Element, or integrate them into a third-party workflow for do-it-yourself library prep.
Elevate is a friendly option for anyone new to NGS. Established NGS labs might choose Elevate to streamline existing workflows and improve quality. Featuring comprehensive solutions with all the quality benefits of a native solution, the Elevate Workflow delivers a well-rounded library prep that is easy to bring in-house.
Charting a Cloudbreak Freestyle™ path
Cloudbreak Freestyle sequencing kits, the latest innovation to ABC, take the freestyle name seriously. Inherently compatible with Elevate and most third-party libraries—meaning, no manual library conversion required—Cloudbreak Freestyle removes lingering limitations to open science up to the outer bounds of your own creativity. This new entry point maximizes ease-of-use and compatibility, letting you choose your own method of NGS involvement.
Open questions meet an open ecosystem
Library prep is the foundation of NGS-based research, the first step you take to getting answers. Embracing the full range of library prep options extracts maximum utility out of a single, ABC-based instrument while creating boundless opportunities to advance your science. Hardware does not need to limit your options.
ABC resists the industry trend of locking labs into a closed ecosystem. Rather, it is designed to stay flexible and keep pace with the rapid pace of genomic innovation. A modular approach to the core chemistry establishes a future-proof foundation for capabilities that start with any library prep and deliver results for any application.
]]>urn:uuid:ef842d90-bc50-4a34-9a65-e2a8f6018e52At AGBT, Element Launches New Tools to Empower Researchers to Shift Their Science2024-02-27T09:35:00-08:002024-02-27T10:01:27-08:00Meredith Ashby, PhDmeredith.ashby@elembio.com
Two years ago at AGBT, Element used the industry’s biggest stage to announce its first product, the AVITI™ System. In 2024, Element showed its ability to innovate not only sequencing but to transform the AVITI into a systems biology platform for the future of biology.
In a series of talks, poster presentations and a workshop on the main stage, Element and collaborators described how the new products launching this year, including Cloudbreak Freestyle, Cloudbreak UltraQ, Trinity, and Teton will help customers streamline their workflow, reach new levels of quality, and shift their science with richer applications.
On both Tuesday and Wednesday mornings, Element welcomed an overflow audience for suite talks on how our technology is improving cytogenetics, sequencing quality, and cloud-based analysis, and extending AVITI’s application capabilities with new products to streamline workflow, reduce costs and generate even more reads.
VP of Informatics Semyon Kruglyak explained how Element is pushing the boundaries on sequencing accuracy by delving into the sources of error in sequencing data. Improvements to accuracy beyond Q30 may be beneficial for certain applications such as the identification of low frequency alleles or the improvement of reference genomes. “We went through a big characterization exercise, …and it turned out that when we looked at the high-quality errors, for the most part, they weren’t being generated via the sequencing chemistry,” Semyon reported. His team’s analysis showed that sample prep and data analysis accounted for 57% and 35% of sequencing error, respectively. “If you want to break through the Q-ceiling, you actually have to address some of those error modes,” he explained. Semyon then detailed the innovative workflow changes the team developed to address different flavors of error to reach a new level of accuracy for Cloudbreak UltraQ, the industry’s first commercial Q50 kit.
June Zhao, Sr. Director of Applications, shared data demonstrating the benefits of workflow innovations including Cloudbreak Freestyle and Expert Mode HD. June showed data from customers already using Expert Mode HD to generate higher throughput with applications where greater sequencing depth adds more value than incremental gains in accuracy. Cloudbreak Freestyle, coming in the first half of the year, eliminates the need for library conversion from nearly all application workflows while preserving data quality, solidifying our position as a seamless next-generation sequencing (NGS) option. June shared Cloudbreak Freestyle data from a range of applications, including methylation, proteomics, microRNA, and RNA sequencing. Together, these new capabilities simplify the customer experience while improving both quality and flexibility.
Delivering high quality data pays off for customers running assays such as OncoTerra from Phase Genomics. Phase CEO Ivan Liachko shared superior data achieved when sequencing on AVITI compared to Illumina and described leaps in cytogenetics made possible with the combination of OncoTerra and AVITI.
At our packed workshop, where organizers scrambled to add more chairs to the room, Molly He, Co-Founder and CEO, described Element’s innovation as “built upon relentless questioning of the status quo and asking what if, what if you can sequence even better, faster and more accurately.”
One of Element’s answers is the forthcoming Trinity assay, coming in 2024 H2. Shawn Levy, Element CSO and SVP of Applications, presented more details and early data demonstrating how this assay will enable researchers to move most of the upfront workflow involved in probe-based targeted sequencing onto the AVITI system, not only reducing hands on time but also significantly reducing probe consumption and improving capture data quality. Trinity will launch with a human exome kit, but Shawn shared a longer-term roadmap that includes customizable probe sets for any research focus area and the capability to tune background whole genome coverage for genotyping by sequencing.
However, the star of the workshop was AVITI24, a powerful tool that consolidates many different instruments and assays into a single box to study systems biology. Element shared further details about how AVITI24 enables detection of multiple analytes in a single assay by combining sequencing and cellular profiling on one instrument. Customers apply cultured, suspended cells directly to an AVITI flow cell with a special surface that enables cell capture. In one 24-hour run, customers can characterize cell morphology, DNA, RNA, proteins, and protein phosphorylation on a single cell basis.
At launch, customers will have access to three panels (Human MAPK/ Apoptosis, Human MAPK/ cell cycle, Human Immune Profiling), each targeting an array of analytes that together will yield a uniquely comprehensive view of a critical cellular pathway on a per cell basis, allowing scientists to make direct observations of how changes in an individual cell’s DNA or culture conditions translates into changes at the RNA, protein, phosphorylation state, and cellular morphology level. Customers who also attended the Wednesday morning suite were treated to an even deeper dive into the technology, presented by Sinan Arslan, Element scientist and AVITI24 project lead, and early access collaborator Dayu Teng, Co-Director of the Whitaker Institute for Biomedical Engineering at UCSD.
Element also announced multiple partnerships at AGBT, including the launch of IDT’s new NGS products designed exclusively for Element’s AVITI System, a collaboration with Twist Bioscience for comprehensive exome workflow for AVITI, and a collaboration with DNAnexus® to advance multi-omics analysis.
Finally, Element showed its playful and vibrant culture in a Willy Wonka-themed “Pure Imagination” party that attracted crowds until the wee hours of the morning. With candy stations, employees in costume, DJ Elevate throwing the dance beats, chocolate cocktails and a fun photo booth, Element’s party made a sweet impression. With such an enticing array of product offerings in 2024, we think Element customers will feel like kids in a candy store.
View Element’s posters and presentations for yourself by visiting our AGBT landing page.
]]>urn:uuid:60505f7b-77f3-41c1-bce0-274fe6c38636Spotlight on Our AVITI™ for All Grant Winner and Gut Microbiomes2024-02-06T10:42:00-08:002024-08-07T12:03:39-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
Innovative genomics solutions require the creativity of scientists and the funding to move research forward at a competitive pace. The inaugural grant in the AVITI Accelerator Grant Program, the 2023 AVITI for All Grant, created the opportunity to advance any research from cancer genomics, plant science, animal science, or metagenomics to any other field of biology. Avidite base chemistry—the unique technology that powers AVITI sequencing—supports a broad spectrum of applications, in turn allowing us to make this opportunity available to virtually any area of research.
For the 2023 AVITI for All Grant, offered in partnership with AVITI service provider AUGenomics, Element Biosciences received many applications reflecting a brilliant diversity of research areas and ideas. From among all the excellent submissions, Element chose a winner who proposed deepening a microbiome study with isolate sequencing while giving undergraduates an opportunity to publish.
Patrick Degnan, PhD, Associate Professor, University of California, Riverside
Exploring complex microbial communities
The winning proposal explores how the gut microbiome mediates the metabolic impact of thiamine supplementation on consumption of a high-fat diet. At University of California, Riverside (UCR), Patrick Degnan seeks to understand how individual species contribute to the overall functioning of complex microbial communities through a combination of metagenomics, isolate cultivation, and molecular genetics. One area of interest is understanding how dietary deficiencies in vitamins such as thiamine (vitamin B1)—a deficiency that occurs even in developed countries—can confer a range of deleterious health effects.
In previous experiments, Degnan and his UCR colleagues have shown that thiamine supplementation has a protective effect for mice fed a soybean oil high fat diet (SO-HFD), reducing weight gain by ~40% over the course of 18 weeks. To explore how thiamine can drive such different metabolic outcomes, his lab is delving more deeply into the mouse fecal microbiomes. Students taking Degnan’s experimental microbiology course have isolated ≥ 1000 anaerobic gut bacterial strains from the fecal pellets of study mice, identifying numerous microbes that are unique to each treatment group.
Sequencing isolated strains
With additional library prep support from Quantabio, the 2023 AVITI for All Grant empowers Degnan and his team to start sequencing isolated strains, providing insights into the functional capacities of microbes that vary in abundance between mice on different diets. Students will conduct assembly analyses and the annotation of sequencing data. The students will also have the opportunity to publish their results as first author in Microbiology Resource Announcements (MRA), an American Society for Microbiology (ASM) journal.
We are excited to support Patrick’s work as both a researcher and an educator, and to find new avenues for continuing the work of making sequencing accessible to all. Helping drive innovation and supporting students is certainly a highlight.
]]>urn:uuid:ee5c3e91-dccd-4e86-9272-af2b81b082d5Cloudbreak™, AVITI™ FIT, and Customer Successes Underline a Banner Year2024-01-23T12:17:00-08:002024-08-07T12:06:31-07:00Jeanene SwansonJeanene.Swanson@elembio.com
Marking the dawn of a new year includes looking back and assessing our accomplishments and lessons learned. And, as it turns out, 2023 was a standout year for Element Biosciences and the Element AVITI™ System. As we continue to innovate on our product offerings and engage with an increasing number of customers and partners, Element looks forward to making 2024 even more productive. We’re proud to share some of what we’ve achieved with an eye on what’s to come in 2024.
Figure 1. AVITI FIT includes Elembio™ Cloud, which extends AVITI functions such as planned runs and consolidates data management in one user-friendly platform.
Committed to constantly innovating
Our unique avidite base chemistry (ABC) offers a completely different next-generation sequencing (NGS) solution. ABC lowers run costs and improves performance through innovations across all fronts of surface chemistry, biochemistry, engineering, and data analysis. A modular foundation ensures extensibility.
Building on our customer-centric model, Element continued to innovate in 2023, launching several new solutions. In 2023, we built out the $200 genome program, launched Cloudbreak enhancements to our ABC technology to reduce run times, increase accuracy, and enable even more applications, and released a suite of products called AVITI FIT for even more flexibility.
With Cloudbreak, customers can scale their experiments and enhance efficiency in multiple ways, including increased sequencing speed with 20% faster run times. AVITI FIT expands on AVITI by offering a suite of lower throughput capabilities, longer read lengths, and individually addressable lanes—all at a low cost.
Earning credibility and gaining influence one AVITI at a time
We know we’re a young company with a new technology—and that we’ve got our work cut out for us when it comes to building trust with our customers. Element has already made significant strides in a competitive and wide-ranging NGS market, gaining international attention and acclaim. Cofounder and CEO Molly He, PhD, was included in the Forbes 50 over 50: Innovation list for 2023. With her help, we have raised more than $400 million from investors.
And did you know that we’ve already sold 100 instruments? In September 2023, Element announced that we had exceeded 100 orders of AVITI. With US and international customers in 25 countries, we’ve expanded our commercial team globally through direct sales and distribution networks. Element has also signed distribution agreements with 11 distributors across the globe. By building interest and delivering on our products, we’ve greatly expanded our reach.
Talking customer success
Not only has Element launched upgrades and continued to expand our reach, we’re also starting to hear success stories from AVITI customers. In a Genetic Engineering and Biotechnology News (GEN)-hosted webinar, “AVITI in Action,” three of our earliest customers—Anoja Perera, Stowers Institute for Medical Research; Adam Majot, PhD, AgriPlex Genomics; and Lutz Froenicke, PhD, University of California, Davis—participated in a panel discussion where they shared insights about AVITI, from purchase to experiment.
Majot said that AVITI FIT stood out as a “great addition,” and pointed to the low- and mid-output sequencing kits. “It’s really nice to have the flexibility to run more specifically for the number of reads you actually expect,” he said. “It’s easier on our servers, easier on our storage.” Froenicke, who runs a core lab, highlighted how useful independent dual flow cells are for faster turnaround and cost-effective scaling, commenting that other sequencers “are too big for us, that would slow us down way too much.” Essentially, AVITI produces runs at a cost comparable to a high-throughput instrument without needing to batch.
The cost savings are undeniable. “I started looking at the data and the cost savings, and I just had to get it,” Perera said. “We are seeing huge cost differences,” estimating about 50% in savings. Element “came up with a sequencing chemistry that is so different from anything else—very, very different—and it works amazingly well,” Froenicke said. “It is very impressive.”
Low cost does not equal low performance, however. Admitting that he was “a little skeptical at first,” the “phenomenal data” was what ultimately won over Majot. “[We] run into a lot of homopolymers and short nucleotide repeats, and it handles those extremely well; and the reliability of the data that we get is quite good ... especially with long stretches of homopolymers,” he said.
Element in the journals—onward and upward
One final note from 2023: we’re starting to see momentum with preprints. Scientists validated the accuracy of our data in multiple studies, most recently one from Andrew Carroll, PhD, and a team at Google in an ABC-focused preprint “Accurate human genome analysis with Element Avidite sequencing.”
Figure 2. Avidites—dye-labeled polymers carrying many identical nucleotides—form the backbone of ABC, labeling and imaging polonies with minimal reagent consumption.
In Asia, customer Burning Rock Dx, a biotechnology company focused on precision oncology, bought several AVITI systems after performing validation work to support research activities. “The data quality of Element gave us a lot of confidence, as we saw improved sequencing accuracy, better genome coverage, lower duplication rates, and better performance in difficult genomic areas such as homopolymer region,” said Chief Technology Officer Joe Zhang, PhD. “This can help us further improve the performance of oncology research products and continue to strengthen our technological leadership.” The preprint outlines a path for reducing the price of comprehensive genomic profiling.
Ready to change your NGS game? Meet us at an upcoming events in 2024 to learn more about how AVITI can kickstart your research.
]]>urn:uuid:c6f821f2-ae70-4a79-9b4c-b6f9374ed58eWhole-Exome Sequencing: An Efficient Path to Understanding Disease2024-01-09T11:34:00-08:002024-08-07T12:08:21-07:00Isaku Tanida, PhDisaku.tanida@elembio.com
The human genome spans about 3 billion base pairs packaged in 23 pairs of chromosomes.1 Approximately 23,500 genes are scattered throughout the genome and bookended by other regions of DNA. Collectively, these genes include about 180,000 protein-coding regions called exons, which comprise the exome.1 Encoded within this human complexity are answers to questions researchers have about myriad health conditions, including inherited disease and cancer. With whole-exome sequencing (WES), they do not have to sequence an entire genome to trace phenotypes and identify disease-related variants.
Figure 1. Where exons fit in the structure of DNA
Comprising only 1% of the genome, exons nonetheless harbor 85% of the genetic mutations known to cause disease.2 By isolating and assessing only exons, researchers extract meaningful data from less DNA to save time and money. WES assesses exons for known and candidate variants, asserting utility for population genetics and research of genetic disease and cancer. The abundance of each variant type is wide-ranging—from tens to millions per genome—so sensitive and accurate sequence detection requires optimization across multiple parameters.
Variant
Approximate Quantity
DNA sequence variants (DSVs)
4 million
Single nucleotide polymorphisms (SNPs)
3.5 million
Nonsynonymous coding SNPs (nsSNPs)
13,500
Loss-of-function (LoF) heterozygous variants
100–120
Variants associated with inherited diseases
50–100
De novo variants
30
Table 1: Abundance of variants in the human genome1
Library prep—Prepare a library from DNA input to access a genome.
Target enrichment—Select the 1% subset of DNA that encodes proteins.
Sequencing—Read and convert the library into files that contain the genome sequence.
Analysis—Gain insight from the sequencing data.
WES is essentially targeted sequencing. Oligonucleotide probes complementary to all exon regions capture relevant DNA for sequencing. Unwanted DNA is washed away. These whole-exome probe panels are readily available through a variety of suppliers. Panels vary in size, hands-on time, automation capability, and compatibility with analysis software and variant databases. When selecting a solution, weighing panel size and completeness against the cost of sequencing required for a panel-appropriate depth of coverage is important.
Figure 2. The WES workflow from DNA extraction through sequencing
Deep insights with less commitment
Arrays and whole-genome sequencing (WGS) have similar goals as exome sequencing but differ in breadth of variation detection within a genome, making WES something of a happy medium:
At one end of the spectrum, arrays detect only variants that are common to many individuals in a population. They also require more input and specialized hardware.
At the other end, WGS reports all nucleotides in the genome, picking up both coding and noncoding regions and mitochondrial DNA (mtDNA).
For certain applications, the high volume of sequencing data that WGS provides is worthwhile. WGS is, for example, a better fit for discovering novel genes and mutations. However, when detecting coding variants is sufficient, WES provides a faster, more cost-efficient approach. As disease-related variants are highly likely to dwell in protein-coding sequences, targeting the exome costs less than WGS while still yielding many useful variants. Moreover, the smaller dataset that WES generates enables faster, more manageable analysis.
Sorting true variants from false
Like any detection method, WES is vulnerable to false positives and false negatives, making sequencing quality extremely important. Incorrect base calls lead to false-positive and false-negative mutations, a problem that higher sequencing quality with fewer errors to begin with mitigates—as does higher coverage to mask low-probability errors. Therefore, higher quality translates to higher accuracy, in turn distinguishing true variants and enabling reduced coverage that can save on costs or increase throughput. Conversely, researchers have the flexibility to sequence to a greater depth.
Uniformity of coverage, another accuracy driver, indicates whether target enrichment captured each sequence at roughly equal rates. Minimally captured target sequences compel additional sequencing runs to ensure adequate representation, a consequence represented as fold-80 base penalty: the amount of extra sequencing reads needed for 80% of the targets to reach mean coverage. A low fold-80 base penalty is desirable because it indicates uniform target representation and cost-effective sequencing. A high fold-80 base penalty wastes reads on well-captured targets and requires more sequencing.
Optimal quality and cost
Avidite base chemistry (ABC) further optimizes the WES workflow by offering a high-quality, low-cost alternative to sequencing-by-synthesis (SBS). ABC, which powers the Element AVITI™ System, delivers superior accuracy across a range of coverages.3,4
A study assessing AVITI performance with Twist Exome 2.0 generated 1.1 billion reads at a sequencing cost of only $45 per exome. AVITI exceeded 90% Q30 and achieved nearly 90% Q40 bases with uniform coverage, efficient depth of coverage, and a low duplication rate. Fold-80 base penalty reached only 1.3, demonstrating efficient sequencing that delivered uniform coverage.
Metric
Value
Assignment rate
97.4%
Coverage > 50x
86.9%
Duplication rate
2.7%
Fold-80 base penalty
1.3
Q30
97%
Q40
86.9%
Read count
1.1 billion
Yield (Gbp)
159.9
Table 2: AVITI delivers high-quality exome data
Figure 3. Fold-80 base penalty results for 8-plex AVITI runs based on Picard HsMetrics (5)
Marian, A.J., “Sequencing Your Genome: What Does It Mean?” Methodist DeBakey Cardiovascular Journal 10, no. 1 (January 2014): 3–6, https://doi.org/10.14797/mdcj-10-1-3.
Arslan, Sinan, Francisco J. Garcia, Minghao Guo, et al., “Sequencing by avidite enables high accuracy with low reagent consumption,” Nature Biotechnology (May 2023): https://doi.org/10.1038/s41587-023-01750-7.
Carroll, Andrew, Alexey Kolesnikov, Daniel E. Cook, et al., “Accurate human genome analysis with Element Avidity sequencing,” bioRxiv (August 2023): https://doi.org/10.1101/2023.08.11.553043.
]]>urn:uuid:69db1363-7cf6-438e-8b0c-36f1efef8876The Five Posts Everyone Read in 20232023-12-18T07:00:00-08:002023-12-18T08:56:25-08:00Chelsea Santoschelsea.santos@elembio.com
We lunched and learned, we watched webinars, we built partnerships, and best of all we got to talk to YOU. We learned about your sequencing challenges, celebrated your successes, and were humbled by the warm reception you gave Cloudbreak™ and AVITI™ FIT. As we bring the year to a close, let’s revisit the blog posts that resonated most with our community in 2023.
5. Bring your own cloud
You asked, we answered: Elembio™ Cloud delivers a much-requested feature that allows remote, real-time run monitoring. As the fifth-ranked post explains, Elembio Cloud features evergreen, ever-growing capabilities that simplify bioinformatics.
Bisulfite samples typically require a sizable PhiX spike-in to increase diversity, making human health studies expensive. Enter avidite base chemistry (ABC), which maintains accuracy without diversity, so you waste fewer reads on PhiX while lowering per-run costs.
Single cell is a star, notching the #3 spot and making a strong case for the ongoing appeal of this versatile sequencing technique. Many people were understandably curious about what Element brings to the single-cell space.
Savings proved a popular topic overall. Our silver-medal post clinches the trend with a walk-through of our $200 Genome program. Shawn Levy, PhD breaks down the pricing for multiple scenarios and explains in plain terms the benefits of this volume-based program.
Agriculture clinches the top spot with a post on low-pass whole-genome sequencing (WGS) and the future of genotyping. This year saw agriculture come to the forefront of genomics, perhaps due to the convergence of climate change and low-cost, end-to-end sequencing poised to replace microarrays—making 2023 a good time to assess options.
]]>urn:uuid:3e15cece-1ebd-4fcb-9c00-6de6eb7acc3eABC™ for NGS: How to Lower Costs and Raise Quality2023-12-04T15:24:00-08:002025-09-04T13:29:23-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
Next-generation sequencing (NGS) has transformed our understanding of biology from human health and disease to plants, animals, and the world of microbes. Past advances fueled a brilliant era of genomics, but core technology gains have since stagnated. Avidite Base Chemistry™ (ABC) is an original sequencing technology that leverages the power of avidites to disrupt establishment models with foundational improvements to cost, quality, and flexibility.
Like sequencing-by-synthesis (SBS), ABC sequencing amplifies and sequences DNA libraries, including those prepared from RNA. Differences in how each technology approaches this function, however, allow NGS applications to evolve beyond SBS.
PCR-free polonies
ABC innovations start with rolling circle amplification (RCA). The flow cell surface captures DNA templates that RCA then replicates and organizes into polonies, where many copies of the template exist as a single concatemer.
RCA improves data quality in two key ways:
Low-binding surface chemistry creates negligible, nonspecific binding and increases the contrast-to-noise ratio (CNR).
Copying only the original DNA template removes a common source of error propagation and data decline: PCR.
Two tasks, two steps
Any polymerase-based method of sequencing must accomplish two tasks:
Detect nucleotide incorporation
Advance the template to the next position
SBS locks these tasks together as a single step, advancing the register when a blocked, dye-labeled nucleotide is added to the growing complementary strand. Covalent incorporation enables a persistent signal for base detection but requires micromolar reagent concentrations to complete the reaction in a reasonable timeframe.
ABC separates these steps, optimizing enzymes and enzyme conditions for each and consuming mere nanomolar reagent concentrations to create stable complexes for base detection.
Avidites and the power of affinity
Avidite refers to the cumulative strength of multiple affinities of noncovalent binding interactions, which is achieved when multivalent ligands bind to multiple sites in a substrate. Avidites—dye-labeled polymers carrying many identical nucleotides—leverage this strength to label and image polonies with minimal reagent consumption.
Although avidites have similar association rates as the dye-labeled monovalent nucleotides of SBS, ABC maintains the advantage: avidites show no disassociation during the > 1 minute needed for base detection, even at nanomolar concentrations. After base detection, buffers wash away the avidites without the DNA scarring that cleaving often leaves.
Figure 1. A detail view of an active site on a polony
Spotlight on homopolymer regions
ABC is not only less expensive than SBS—it is also more accurate. The accuracy gain is particularly prominent in homopolymer regions, which maintain low error rates pre- and post-homopolymer. Element researchers attribute the higher accuracy to RCA, an engineered high-fidelity polymerase, and two additional factors:
Synergistic binding of multiple nucleotides on a single avidite makes sure that only the correct avidite binds to a polony.
A binding disadvantage for out-of-phase DNA copies that are inside the polony and lack other out-of-phase neighbors to serve as avidite substrates.
Figure 2. Mismatch percentages before and after passing through > 12 bp homopolymers
Novel technology with unexplored potential
Avidites enable a modular design that presents clear paths forward for further innovation and improvement, making ABC a highly extensible technology. The polymer core, number and choice of dyes, and length and structure of nucleotide linkers are set up for parallel optimization that increases signal, decreases cycle times, lowers reagent concentrations, and tunes other parameters to neatly align to different applications.
For example, a novel polymerase and optimized reagent formulation have already enabled 2 x 300 sequencing. The results are excellent. Cloudbreak™, which accelerates run times by 20% and enables linear library loading, is another example.
Publication and proof-of-concept
Entwining low cost and high quality, ABC takes a first-principals approach to stretch beyond the limits of SBS and empower more labs to do more science. In a seminal publication, Element researchers describe the kinetics of ABC in detail, including proof-of-concept data from human whole-genome sequencing (WGS) and single-cell sequencing. Datasets provide further visibility into ABC performance.
]]>urn:uuid:391f6829-9ea9-4fe2-9cc1-b07b01dff074Reducing the Price of Comprehensive Genomic Profiling2023-11-27T16:00:00-08:002024-10-02T07:34:16-07:00Brian Coullahanbrian.coullahan@elembio.com
An ability to simultaneously interrogate multiple biomarkers from samples with high heterogeneity—as is the case with tumor samples—has driven rapid uptake of next-generation sequencing (NGS) across the field of oncology research. Therapies that leverage your own immune system and compounds that target specific oncogenes have also shifted the field. The development of novel methods to identify the most effective treatment regimen based on someone’s unique tumor profile are an opportune result. Although research into these companion diagnostics brings great promise to increased adoption of NGS-enabled multigene testing, the cost of sequencing larger comprehensive genomic profiling (CGP) panels hinders adoption and stifles development.
A team at Burning Rock Dx sequenced libraries prepared with their CGP panel, OncoScreen Plus, on the Element AVITI™ System and Illumina NovaSeq 6000 System and compare the results in a preprint. Burning Rock had already validated NovaSeq, so the goal was to determine whether the team benefitted from the flexibility and improved costs of AVITI without impacting results.
Figure 1. OncoScreen Plus and AVITI unite to deliver an efficient, high-quality CGP workflow.
Two studies, comparable results
The team performed two research studies to ensure that all reported results were comparable. Each study comprised a comprehensive analysis of known and novel CNVs, SNPs, and indels and complex biomarkers such as microsatellite instability (MSI) and tumor mutation burden (TMB).
Figure 2. CGP leverages a single NGS assay to assess hundreds of cancer genes.
The first study leveraged a set of contrived control samples—which allow greater control of large datasets—from a cell line harboring well-characterized variants at known frequencies from ~1% to > 90%. Diluting the sample with a germline control at factors of 1/2, 1/4, 1/8, and 1/16 generated a dataset comprised of hundreds of variant/allele frequency combinations, allowing Burning Rock to determine the concordance levels in detection and relative predicted frequency for both systems. Results show high concordance, detecting the 69 known variants in the control sample and 353 variants detected across the targeted region of the panel.
The second study focused on comparing the results of real-world tumor samples from FFPE blocks. Eight FFPE controls harboring a set of known variants were sequenced on AVITI and NovaSeq. Results indicated nearly identical performance with correlation coefficients > 0.9 for five of the eight samples. Only one sample resulted in 0.75 because of an outlier data point. Additionally, all 35 CNV calls that NovaSeq detected were identified and given the same copy number call as AVITI.
AVITI amplification advantages
In addition to detecting genomic alternations, the OncoScreen Plus assay generates results for complex biomarkers, allowing further comparison across both systems. Unsurprisingly, from both tumor mutation burden and microsatellite instability calculations, all samples in both studies produced highly concordant results.
Remarkably, the authors note a consistently higher average coverage of short tandem repeats (STRs) in AVITI data versus NovaSeq. Similar results observed in Element Biosciences studies are primarily attributed to reduced error propagation during amplification. NovaSeq and most other systems built on sequencing-by-synthesis (SBS) chemistry rely on PCR-based clonal amplification methods such as bridge amplification. AVITI employs rolling circle amplification (RCA) to innovate significant accuracy advantages. RCA continuously copies the original template to improve quality across regions that typically result in PCR stutter, such as homopolymers, STRs, and other repeat elements.
Observation of optical duplicates revealed an additional RCA advantage, with AVITI showcasing fewer optical duplicates. The unbound nature of bridge amplification can lead to the migration of template molecules across regions of the flow cell and colony duplication. The duplicates are later removed from results but can still account for a significant amount of data and reduce usable results. In this case, the Burning Rock team removed ~5–10% of Illumina reads. The tethered Element approach prevents optical duplication and permits almost no data loss.
A trailblazing CGP comparison
The Burning Rock study is the first study to compare CGP results on Element and Illumina systems, and it will not be the last. Given the significant cost savings and flexibility gains to reduce batching constraints, the field is anxious to explore alternatives to current sequencing solutions.
Moreover, the authors note that transitioning their workflows from Illumina to AVITI was virtually seamless and they ran identical libraries on both systems. This assessment includes the analysis pipeline, which was unmodified for analysis of AVITI results. The team expects that further optimization of analysis pipelines for AVITI data can correspondingly push quality even further.
]]>urn:uuid:b8a12151-98f1-41cf-9fc0-3dbcc6cbdfd52 x 300 Sequencing: All the Reads You Need for Metagenomics2023-11-20T06:00:00-08:002023-11-27T16:17:09-08:00Terrence Gelineauterrence.gelineau@elembio.com
Since Antonie van Leeuwenhoek first peered into a microscope to study microorganisms in the 17th century, microbiology has made the remarkably diverse, unseen world around us visible. The advent of next-generation sequencing (NGS) introduced a culture-free solution that revealed the true diversity of microbial communities living on, in, and around us, sparking deeper exploration into the composition and function of these important ecosystems.
Figure 1. Metagenomics applies genomics methods to study the composition and function of microbe communities. Formally used to describe only shotgun whole-genome sequencing (WGS), metagenomics increasingly describes multiple methods.
Tools to assess microbes and their environments
A range of methods cultivate our understanding of the structure and function of microbial communities. The most comprehensive method is shotgun sequencing, which samples all genes in a community to understand its functional potential. Shotgun sequencing can answer questions about which fuel sources a community can consume, metabolites it can produce, and antibiotics it can resist. Metatranscriptomics enhances this assessment with a snapshot of ongoing activity in an environment at the time of sampling.
Together, metagenomics and metatranscriptomics form a formidable tool for understanding the influence a microbiome has on an environment. However, the throughput required for metagenome sequencing is often high, especially for soil and other complex communities. Ascertaining whether additional sequencing might pick up additional genes that are low abundance but functionally important is difficult and expensive.
An alternative, more economical approach is to focus on community composition via targeted sequencing. High-level community functions can be inferred from known information about the biology of member species. Moreover, the impacts of environmental perturbations or stratification are observable across many samples with far fewer sequencing reads. Even as sequencing costs have retreated, targeted sequencing remains the most popular metagenomics approach.
Targeted sequencing for metagenome profiling
The 16S rRNA gene is ideal for profiling composition. The gene is sufficiently conserved across bacterial clades for reliable amplification with universal primers, and with nine variable regions (V1–V9) contains enough sequence diversity to distinguish bacteria at the species or strain levels.
Figure 2. Example targeting of the V3–V4 region in the 16S rRNA gene. Blue indicates highly conserved regions and yellow marks variable regions.
The sequence diversity of each variable region can differ between bacterial clades, so community profiling requires various amplified gene fragments. The common use of V3–V4 amplicons, which span longer gene fragments, requires up to 2 x 300 cycles of paired-end sequencing to produce a consensus sequence. At the ends of these long reads, sequencing-by-synthesis (SBS) accuracy declines, which can erode quality in the read overlap region.
Here, Element picks up where SBS falls short. Our chemistry generates 2 x 300 reads that maintain Q30–Q40+ quality throughout Read 1 and Read 2 to deliver high-confidence data for characterizing complex biological systems.
Figure 3. Sequencing human control DNA HG001 delivers > Q40 scores throughout Read 1 and persists with > Q30 through the end of Read 2, outperforming SBS. The Elevate™ Workflow and Adept™ Workflow generated PCR-free libraries from HG001. Four runs at Element Biosciences sequenced the Elevate library. A service provider sequenced the Illumina library two times.
]]>urn:uuid:3c02141f-92db-496b-b358-b84899bac90cElement Spotlights Rare Disease, Gene Expression, and Cytogenomics at ASHG 20232023-11-13T16:48:00-08:002023-11-14T12:53:36-08:00Kristi Heimkristi.heim@elembio.com
Element Biosciences made a strong showing at ASHG 2023 in Washington, D.C. progressing through a series of announcements and presentations, including a workshop detailing variant calling with the Element AVITI™ System and rare disease. A lively cocktail party capped off events with hundreds of clinking glasses and a bevy of some of the brightest minds in genomics.
Figure 1. The Element team gets ready to address your sequencing challenges.
Strong ecosystem through partnerships
Element chose ASHG to debut the AVITI Accelerator Grant Program, starting with an inaugural 2023 AVITI for All Grant in partnership with AUGenomics, a trusted AVITI service provider. We also announced a partnership with QIAGEN to advance genomic applications through end-to-end sequencing workflows that integrate QIAGEN panels and bioinformatics with AVITI.
More generally, partnerships emerged as an important theme. QIAGEN not only announced our partnership—they delivered an in-booth presentation. Element was also thrilled to welcome additional partners, Revvity and Agilent, to our booth as presenters. Through these ecosystem partnerships, Element looks forward to putting next-generation sequencing (NGS) within reach of any lab.
Joint learnings on rare disease
In collaboration with TGen, part of City of Hope, Element presented the results of an AVITI-based research study on rare disease that identified the likely genetic causes of neurological disorders in children. Employing a novel design that sequenced the parents at half the child’s coverage allowed the entire trio to be sequenced on one flow cell while retaining the ability to identify de novo variants.
FYR Diagnostics performed the whole-genome sequencing, which took fewer than 48 hours to complete and cost $1680 per trio. Since the study, the introduction of Cloudbreak™ chemistry has further reduced run time to 38 hours.
Figure 2. Results for HG001 demonstrate improvement with increased insert length and pangenome reference. Improved accuracy and alignment reduce total errors and increase F1 scores.*Figure 3. Semyon Kruglyak, PhD, VP, Informatics, Element Biosciences, presents a poster displaying the study design and results (left). Keri Ramsey, BSN, RN, Clinical Co-Director, Center for Rare Childhood Disorders, TGen, co-presents with Kruglyak in our workshop covering the study in more detail (right).
Gene expression and cytogenomics with Element data
Patrick Kennedy from the La Jolla Institute for Immunology spotlighted Element in a platform talk, describing how he leveraged AVITI to develop a new method based on single-cell sequencing to empirically determine how genetic mutations impact T-cell signaling by changing protein phosphorylation events. The method applies CRISPR screens to perform a modification, followed by an assessment of the impact on gene expression within pathways of interest.
A poster by Phase Genomics and Fred Hutch Cancer Center also featured AVITI data. Centering on next-generation cytogenomics, the poster addressed the potential of Hi-C DNA sequencing to consolidate data in a single workflow—currently, multiple distinct assays are a limitation. Workflow consolidation is poised to simplify lab operations, reduce costs, and use limited samples more efficiently. Additionally, pairing OncoTerra with AVITI can speed turnaround time by allowing labs to batch fewer samples without exceeding cost targets.
Until next year …
Thank you to everyone who dropped by our booth, checked out our posters, joined us for lunch or cocktails, or attended one of our presentations. If you missed the event, we invite you to catch up on-demand. We look forward to seeing you at ASHG 2024.
]]>urn:uuid:2d5a4c20-62ba-4699-824e-2e816316567fHow Element Got to the $200 Genome: Shawn Levy Explains2023-10-09T07:30:00-07:002023-12-01T10:08:20-08:00Shawn Levy, PhDshawn.levy@elembio.com
Different sequencing applications have different needs. Metagenome taxonomic profiling, for example, benefits from low error rates and has variable output requirements, while whole-genome sequencing (WGS) benefits from high accuracy at high throughput. What these applications did not have in common—until now—is a workable price point, one that adapts to low, medium, and high throughput.
Recent advancements in sequencing have reduced prices at all experiment scales without needing to batch large numbers of samples. A natural extension of this innovation is the $200 Genome, a program that decreases the cost of a 2 x 150 run to drive WGS and WGS-equivalent applications such as counting and single-cell, enabling higher volume sequencing for mid-throughput labs.
Eight-fold lower sequencing costs compared to NextSeq 2000
A run under the $200 Genome program outputs 300–330 Gb of data, sequencing three genomes or equivalent on one flow cell at 30–35x coverage. At this output, cost is as low as $200 per genome or $2 per gigabase. Essentially, this program presents an opportunity for a benchtop sequencer to reduce costs by 8-fold compared to competitor systems.* The more you sequence, the more you save.
Figure 1: The $200 Genome Program synchronously lowers the price per kit and price per genome, driving accessibility and value for higher volumes.
Starting with as few as ~1000 genomes annually and increasing up to ~6000, all outputs benefit from instrument redundancy, flexibility, and value, with maximum savings at the high end of the range. Delivering the same value as larger platforms with commitments of tens of thousands of genomes per year, the $200 Genome scales these savings for thousands of genomes per year.
A look at more specific numbers and use cases
Threshold usage of 100 flow cells per quarter equals an effective sequencing kit price of $1380, which is slightly lower than list price. Ramping up to moderate usage allows more instruments and a big drop in effective kit price, down to $767, or less than half the list price. Maximum usage, which maxes out at ≥ 230 flow cells per quarter, ups the instrument count to 3–5 and achieves the $200 genome. In pure dollars and cents, this threshold translates to an annual kit commitment of only ~$500,000. Multiple instruments help achieve the highest value with a bonus of greater flexibility, particularly for labs sequencing genomes alongside other applications.
Table 1: Example use cases show savings at every level
Usage
Flow Cells per Quarter
Genomes per Quarter
Effective Kit Price ($)
Per-Genome Price ($)
AVITI Systems
Threshold
100
300
1380
460
2
Moderate
180
540
767
255
3–4
Maximum
≥ 230
≥ 690
600
200
3–5
For perspective on instrument amortization, sequencing genomes under the usual terms with an AVITI System and service contract adds ~$72 per genome. That number still represents tremendous per-genome value with only moderate usage. For genomes and genome equivalents, the $200 Genome drives accessibility and flexibility for high-throughput single-cell and counting applications, cell-free DNA (cfDNA), and other conditions that require tremendous data output, anywhere from tens of thousands of gigabases to the low hundreds of thousands.
Threshold usage of the program still drives exceptional value. At 1380 genomes per year, for example, it is a $400 genome program. As you exceed 2760 genomes annually, the $200 genome becomes accessible to as many genomes as you can run, depending on the number of available instruments. Although using fewer kits raises the effective kit price, it remains a significant discount relative to list pricing and competitors.
Exceptional value for high volume
The $200 Genome program empowers moderate throughput labs, offering significant benefits to labs with enough genomes. More specifically, labs without the throughput or ability to batch hundreds of samples per run still deserve the flexibility of daily run starts and rapid trio sequencing. Labs sequencing even modest amounts of genomes in a year (in the low thousands) no longer need a NovaSeq—this program competes favorably. For day-to-day sequencing that falls outside the $200 Genome sweet spot, Element continues to offer flexibility, value, and performance.
* Based on equivalent NextSeq 2000 P3 data at list price. Comparison does not include preparation, analysis, and warranty costs.
]]>urn:uuid:8b354d18-8dcf-40c2-98e6-4bf5e48768c3Quantification of Circular Adept™ Libraries Made Simple2023-09-27T16:09:00-07:002023-09-29T11:51:06-07:00Terrence Gelineauterrence.gelineau@elembio.com
The Element AVITI™ System uses rolling circle amplification (RCA) for polony generation to decrease error rates caused by other amplification strategies. RCA requires circular library to serve as a template:
Circular libraries are single-stranded, so the recommendation is to quantify them using qPCR. To support customers who prefer Qubit quantification due to speed and ease of use, Element Biosciences maintains a technical note on how to quantify circular libraries using Qubit.
Understanding the qualities of both qPCR and Qubit when determining your routine quantification is essential. qPCR is an accurate method for measuring library concentrations, and is sensitive to lower concentrations. When using the primers supplied in the Adept Library Compatibility Kit v1.1, qPCR detects only circularized library by priming off the outer adapters and targeting the ligation junction of the circularized library during qPCR (Figure 1A).
Using Qubit or other fluorometric quantification methods requires reagents designed for single-stranded DNA (ssDNA). Qubit quantifies any linear DNA contaminating a post-circularization library, including incomplete library fragments, library that did not circularize, primer dimers, splint oligos, and partially digested DNA fragments (Figure 1B). This catch-all quantification can overestimate the library concentration. Furthermore, Qubit cannot detect whether a library has successfully circularized.
Figure 1. Adept qPCR targets (A) and DNA fragments that Qubit might quantify (B)
Secondary structures in single-stranded circular libraries can overestimate the library concentration, making heat-denaturation necessary before quantification. The technical note documents the procedure. After completing the procedure, you can follow manufacturer instructions to Qubit-quantify the library.
Validating your quantification method of choice
Crucially, validating both quantification methods before routinely quantifying libraries using Qubit verifies that Qubit results are as expected for the library type. A linear relationship exists between libraries quantified using qPCR versus Qubit (Figure 2). Because this relationship is variable, labs might choose to verify Qubit with the libraries they commonly use.
Figure 2. Relationship between qPCR- and Qubit-quantified libraries based on linear input for the Adept Workflow. Qubit is not sensitive enough to quantify lower concentrations.
Even after validating Qubit, retaining the ability to quantify with qPCR is important. qPCR results are more accurate than Qubit results and can be used to verify Qubit results, optimize concentrations for new libraries, and troubleshoot unexpected results.
Tracking data about library quality control (QC) and quantification while optimizing Qubit quantifications is helpful. To assist with characterizing the relationship between quantification methods and optimize loading concentration, track the following information:
Sample type and library prep method
Linear library QC data, such as quantification results and Bioanalyzer or TapeStation traces
Adept Workflow input and output concentrations
qPCR and Qubit quantifications of circularized libraries
The number of polonies the sequencing run generates
Troubleshooting ligation and digestion issues on Qubit-quantified libraries is accomplished through qPCR, Bioanalyzer, or a fragment analyzer. TapeStation is possible, but it does not always detect incomplete digestion. Figures 3 and 4 provide examples of ligation failure and incomplete digestion for qPCR.
Figure 3. qPCR quantification depicting example ligation and digestion failuresFigure 4. Bioanalyzer traces depicting example ligation failure (A) and an incomplete digestion (B)
Enabling Qubit quantification for Adept libraries is one of many ways that Element is working to make our industry leading sequencing technology easy to use and accessible to all. To learn more about how the AVITI can help your science now and in the future, contact us here.
]]>urn:uuid:250d2f9c-e929-4ac1-940c-223cb22780b5AVITI™ Genomic Analysis on AWS – Simple, Powerful, and Cost-Effective Cloud Computing2023-08-02T10:28:00-07:002023-08-03T12:12:12-07:00Francisco Garciafgarcia@elembio.com
By: Francisco Garcia, Rosi Bajari, Claudia Dennler, Max Mass, Bryan Lajoie
Sequencing with Element on the AVITI™ System
Genomic analysis is playing an increasingly pivotal role in both research and clinical operations. To achieve efficient and cost-effective sample-to-answer workflows, labs face the challenge of building a cross-functional team with expertise in DNA and RNA sequencing, informatics, cloud computing, and IT. This is particularly daunting for labs that are new to sequencing.
While the AVITI System provides significantly lower run costs than other benchtop sequencing systems, some customers remain daunted by the perceived complexity and cost of analyzing the generated data. In this blog post, we show how Element Biosciences has partnered with Amazon Omics to provide customers with an analysis solution that is fast, flexible, powerful, and very cost-effective using ready-to-run Bases2Fastq workflows.
From Sample to Answer: A Journey in Sequencing
Sequencing data analysis is separated into primary, secondary, and tertiary analysis. Secondary analysis includes three stages, demultiplexing, data transposition, and genomic analysis.
Primary analysis involves processing the signal generated by sequencing instruments to generate base calls and associated quality scores. The AVITI uses an on-board FPGA for primary analysis to accommodate the high data rates produced during sequencing. In fact, the data rate is so high that only the processed data reach the on-board CPU.
The first part of secondary analysis is demultiplexing. With mid-throughput systems like the AVITI, customers typically pool multiple samples together on one flow cell, tagging each with a unique molecular barcode. Demultiplexing identifies the data associated with each barcode.
Transposition, the second part of secondary analysis, involves taking the data that was generated per-cycle and saving it as short read fragments in FASTQ format.
The third stage of secondary analysis, genomic analysis, depends on the intended sequencing application. For DNA resequencing applications, the short reads are aligned to a reference genome. Differences between the given sample and the reference are identified as genomic variants.
Tertiary analysis involves the interpretation of those variants to determine biological significance.
The AVITI can be configured to stream base calls, quality scores, and run metrics as they are generated directly to a customer’s S3 bucket on AWS. Subsequent analysis can be performed immediately without the need to copy to an intermediate location or export data from a managed and costly subscription-based service. With our upcoming fall release, customers will be able to seamlessly configure the automatic launch of Element’s Bases2Fastq Ready2RunWorkflow via Amazon Omics in their own AWS account as soon as their sequencing run completes.
This configuration takes minutes, uses a secure AWS IAM role and External ID, and can apply to all sequencing runs in a customer's account or to a subset of sequencing runs streaming to a particular S3 bucket. Once the execution is completed, Elembio™ Cloud complements Amazon Omics with execution records and the ability to visualize QC statistics in an interactive report.
From run start to analysis completion, customers have full control of their own data in their account. Users can optionally allow Element access to run metadata so that QC analysis can be presented in Elembio Cloud.
Analysis with the Element AVITI and the Omics Ecosystem
Once FASTQs are generated, users can take advantage of the elasticity of Amazon Omics to trigger subsequent secondary analysis for each sample in parallel. A run setup in Elembio Cloud can be configured to launch independent per-sample analysis workflows automatically. For example, one flow cell run comprised of three ~30x human genomes can be configured to launch downstream secondary analysis tools on independent instances as soon as demultiplexing is complete. Because the analyses are fully parallelized, the time to compute all samples is the same as it would be for a single sample.
Data transfer occurs during sequencing.
Demultiplexing with Bases2Fastq takes ~1 hour.
Sentieon DNAscope analysis, included as a 6-month free trial with an AVITI purchase, takes ~35 minutes for an AVITI 30X genome in AWS.
Fully processed human genome data, from base calls to FASTQ to BAM to VCF (variant calls), is available within 2 hours of run completion.
Cost Effective Base Calling with Bases2Fastq on Amazon Omics
So, how much does this analysis really cost? While there has been alarming messaging in the press that analysis costs are greater than sequencing costs, in truth the cost of cloud analysis is only a very small fraction of a sequencing budget. Let's delve into the actual numbers.
For a custom AWS setup, running Bases2Fastq on a m5.12x (48cpu/192g) instance (using spot) with a single attached 800G ebs-auto-scaling gp3 volume, it takes less than an hour to generate FASTQs for a typical 300G AVITI run. However, this setup requires substantial configuration time and ongoing management by a bioinformatics professional, resulting in potentially significant labor costs.
In contrast, utilizing Amazon Omics and the Element-managed Ready2Run Bases2Fastq workflow offers an easier and more cost-effective solution.
FASTQ generation costs approximately $3 for a 300G 2x150 run.
Running DNAscope on a 30x AVITI genome costs approximately $1.60 per sample.
For three pooled 30x human whole-genome sequencing samples on a single flow cell, the total cost per sample is ~$2.60 per sample: $3 per flow cell/ 3 samples for demultiplexing and $1.60 per sample for alignment and variant calling.
The cost per flow cell is $7.80 per flow cell, amounting to less than 1% of the cost of the flow cell itself.
From the perspectives of turnaround time, ease of use, and flexibility, utilizing Omics for genomic data analysis emerges as a significantly superior option compared to local operations. Furthermore, the cost per sample is negligible, making it an exceptionally compelling choice for users of Element Biosciences' AVITI sequencing platform.
To get started with Element Bioscience’s Bases2Fastq Ready2Run workflows, visit the Amazon Omics console.
]]>urn:uuid:19fedb99-e093-4a3f-9980-9b2383f51440Introducing the Element Biosciences Elembio™ Cloud: Remote run monitoring and simplified cloud-based informatics support2023-06-21T17:02:00-07:002023-07-01T20:56:18-07:00Rosi Bajarirosi.bajari@elembio.com
Element is excited to announce the launch of Elembio Cloud, the online platform for monitoring sequencing runs, visualizing run data, and managing your Element account features. Functioning as a direct extension of the Element AVITI™ System, Elembio Cloud enables easily viewed run performance and progress with continuous, real-time updates and access to a rich set of QC metrics explorable in an intuitive web platform. Cloud integration and account management makes adding integrations, planning subsequent runs, and managing your Element sequencing data and team a seamless effort.
Remote Run Monitoring on the AVITI System supports a wide array of sequencing applications - low pass WGS, single cell sequencing, WES, methylSeq, and long read sequencing with LoopSeq™ just to name a few. With multiple instruments and team members, repeated lab check-ins to keep track of multi-application or multi-flow cell sequencing projects can be inefficient. Leveraging remote run monitoring, connected AVITI systems automatically synchronize with Elembio Cloud to provide a rich view of sequencing runs for everyone on the team in an accessible online platform.
Track run status sorted by instrument from anywhere to work more efficiently.
View QC metrics including Q-Scoring, PhiX Error Rate.
Review early demultiplexing to plan your next run while the current one is still underway.
Monitor instrument availability remotely to start subsequent runs promptly.
Remote run
monitoring helps your sequencing team manage lab activities without the need to
be in the lab.
Your Data, Your Choice of Storage and Integration Partners
Genomic data can contain personal and potentially identifying details, and Element recognizes the need for data to be well secured and meet your data storage and analysis requirements. Elembio Cloud allows users to integrate, verify, and manage their connected AVITI fleet so that all data are kept within a cloud environment that you own and manage, not Element. Built-in flexibility of cloud storage partners, including AWS S3 and Google Storage, allows AVITI OS to securely stream source data directly to storage locations with no intermediary steps. Data management in your bucket of choice streamlines swift entry into secondary data analysis.
Streamlining FASTQ Generation with Industry Partners and Cloud Informatics
Converting bases files to FASTQ files is the first step in your cloud informatics pipeline, and it can be done with our Bases2Fastq software. To simplify analysis pipelines, Bases2Fastq supports key features including UMIs, FASTQ generation for all reads (R1, R2, I1, I2, UMI), 0-performance cost adapter trimming sensitive to indels and short adapters, and smart detection of index and adapter sequences. Bases2Fastq acts as an all-in-one software to optimize FASTQ generation and reduce required preprocessing steps before secondary analysis.
To simplify the process of FASTQ generation, Elembio Cloud is partnering with leading informatics providers to create analysis flows that can be leveraged with little to no informatics setup. The recently launched AWS Omics platform is an early partner, featuring a federated Bases2Fastq Ready2Run workflow that brings the compute to your data, in your account. Using Amazon Omics, Bases2Fastq can be run in roughly an hour and a half with minimal setup needed. We additionally support launching Bases2Fastq on Nextflow through the community curated bioinformatics pipeline resource nf-core.
One Stop Shop for Account Management
We know it’s frustrating to have to interact with multiple platforms to manage your sequencing activities. With Elembio Cloud, Element introduces a one-stop shop for your account management. Create your Team and invite your team members to join, or connect to your organization’s secured single-sign-on authentication platform.
Elembio Cloud is an exciting new platform to support all your sequencing operations seamlessly, and this is only the beginning.
Curious about how it all looks?
Check-out our demo site, where you can login with the demo user (username: demo@example.com password: DemoUser!) to experience the platform.
]]>urn:uuid:179bfc73-55f4-4c9a-8be4-ba068e5fed44Cloudbreak™ is here with faster runtimes, simplified workflow, and higher accuracy2023-06-15T08:56:00-07:002024-08-07T13:01:29-07:00Isaku Tanida, PhDisaku.tanida@elembio.com
The AVITI™ System just got even better with the launch of Element Biosciences’ Cloudbreak chemistry, an advanced version of the AVITI System’s avidite sequencing technology.
Cloudbreak features:
20% faster run times across read lengths.
Early indexing for real-time sequencing run management.
Improved data quality at the end of each read for greater accuracy.
No library conversion for the native Elevate™ library prep.
Next-generation sequencing (NGS) has revolutionized the field of genomics, empowering researchers to confront complex scientific questions. But despite these advances, broader access to sequencing through benchtop scale systems has been limited, largely due to high costs. This has driven researchers toward factory‑scale sequencing or alternative platforms that promise savings yet come with significant trade-offs including extended TATs and/or poor data quality.
To overcome these limitations and drive more science, Element Biosciences reimagined the core components of NGS to produce the AVITI System, a benchtop platform designed to provide broad access to the genomics ecosystem. Avidite sequencing forms the core of AVITI’s disruptive design. It is readily compatible across leading applications and technologies while delivering superior data quality at low cost.
The introduction of Cloudbreak chemistry advances the core benefits of avidite sequencing by increasing AVITI’s sequencing speed and workflow efficiency. In only 38 hours, two PE 150 assays with indexing can be run in parallel to produce up to 600 Gb of data and 2 billion reads. For shorter read length assays typically used in RNA-seq and scRNA-seq, users can similarly achieve 2B high quality reads daily.
With Cloudbreak, sequencing speed dramatically improves the daily and weekly scheduling flexibility that users need to run their experiments. Specifically, for short read length assays Cloudbreak run times can push AVITI data outputs from 6-7 billion reads per week to upwards of 10 billion reads per week while comfortably maintaining an 8 to 9-hour workday.
The new index-first run format means AVITI sequences indexes before the DNA insert, allowing for early demultiplexing onboard the instrument. This provides users fast access to the index assignment percentages of each sample in a pooled run. When shorter run times are combined with this early indexing feature, users can manage their flow cells in real time, deciding whether to cancel and re-start a new flow cell if needed , or continue the run and pool select samples on a second flow cell to compensate. This ability to work in real time, using the unique flexibility features of the instrument, saves time that would have been otherwise lost while waiting for a run to fully complete.
An exciting sign of more advancements to come, Cloudbreak also improves the efficiency of preparing libraries for AVITI by eliminating the conversion process from its native Elevate library prep workflow. With Cloudbreak, any Elevate library can now be loaded directly onto the AVITI System after quantification without first requiring library circularization on the bench. Circularization is critical to avidite sequencing accuracy and still occurs, but Cloudbreak moves this step onto the flow cell with Elevate libraries. Best of all, moving the previously manual step onto the instrument adds no time to the run, making Elevate the fastest end to end workflow you can experience on AVITI from library prep to result. We look forward to adding simplification options to the Adept workflow in the near future as well.
In summary, the AVITI System is a true breakthrough in benchtop sequencing, providing superior advantages over its competition in data quality, cost, and flexibility. By adding Cloudbreak speed and workflow enhancements, AVITI further separates itself from the pack by building on its leading resume of capabilities. For more information on how AVITI with Cloudbreak is the perfect sequencing solution for you, please download our new data sets and specification sheets or contact us directly for more information at www.elementbiosciences.com.
]]>urn:uuid:8ce39d46-139d-4a51-8901-3b8962c95b59Single-cell sequencing and the Element AVITI™ System: a perfect pairing 2023-05-01T00:00:00-07:002023-07-31T17:06:41-07:00Isaku Tanida, PhDisaku.tanida@elembio.com
Single-cell sequencing is a powerful technique for revealing hidden complexity within cell populations. The technique has found widespread adoption in fields such as immunology, neuroscience, developmental biology, and cancer research, where small populations of cells with unique gene expression profiles can have profound impacts on entire systems. In single-cell experiments, it is important to choose tools that work together efficiently and cost-effectively to enable the in-depth analysis often needed to generate new insights into human disease or identify new targets for therapeutic interventions. Single-cell sequencing requires at least two paired tools: a system for attaching barcodes to RNA by cell of origin and a DNA sequencing platform to read out the tagged gene expression data.
Choosing a single-cell barcoding platform
Today, scientists have multiple options for efficient cellular barcoding of RNA. All single-cell platforms pursue a basic strategy of isolating and lysing individual cells, then applying reverse transcription to generate cDNA while adding a cell-specific barcode and often a molecule-specific UMI. After barcoding, cDNA from the processed cells is pooled for NGS library preparation and sequencing. The details of the general strategy vary by platform, resulting in differing strengths and limitations. The two most commercially established platforms are the 10X Genomics Chromium and the BD Rhapsody.
The 10X Genomics Chromium System isolates individual cells within nanoliter-scale Gel-In-Bead Emulsions using a microfluidics system. Each droplet contains, ideally, one cell and one bead decorated with many copies of a single barcode. The droplets serve as reaction chambers for reverse transcription (RT).
The BD Rhapsody isolates cells in picoliter wells using gravity deposition. A combinatorial library of barcode-carrying magnetic mRNA-capture beads, sized to optimize the deposition of a single bead per well, is delivered via microfluidics. Once cells are lysed, mRNA binds to the beads and the beads are pooled for barcoding and reverse transcription steps.
Both systems are high-throughput, have similar runtimes, and similar cost per cell for a standard gene expression assay. The 10X Chromium is widely available, simple to use, and provides the largest menu of supported assays. The BD Rhapsody has gentler cell handling, a lower multiplet rate, and the ability to QC runs before investing in sequencing. With differing advantages, the decision on which platform to use will depend on a range of investigator and experiment specific factors.
Simplify your single-cell sequencing logistics
The sequencing portion of the single-cell workflow is an important factor in the success of the overall experiment as systems vary significantly in quality, flexibility, turnaround time, and of course cost. The AVITI DNA sequencing system fits ideally within the workflow requirements of the leading single-cell technologies.
The AVITI System throughput aligns neatly with sample outputs from both leading and emerging single-cell technologies.
High sequencing quality assures assay sensitivity to low expressed genes and high yield of genes per barcode.
Two fully independent 1 billion-read flow cells enable multiple independent run-starts daily for unrivaled sequencer access.
Industry leading cost of $0.60 - $1 per million reads and guaranteed reagent costs for the life of your instrument lets you do more science for your budget.
Platform flexibility is also important. But what is meant by flexibility? A flexible system can accommodate the varying throughput requirements of an ever-widening range of single-cell assays. With partitioned flow cells, cell numbers and read depth requirements may not fit neatly into strictly bucketed lanes, leading to Tetris-like sample planning and inefficient usage that increases per cell costs.
Figure 1. Dual independent flow cells allow you to sequence multiple experiment, using multiple applications, with minimal waiting or coordinating among users.
Flexibility can also mean the ability to run on-demand in a shared system environment. Indeed, one of the primary draws of a benchtop system is avoiding the delays associated with queueing for ultrahigh-throughput, batched runs. Unlike other benchtop platforms, the AVITI System’s dual flow cells can be run completely independently, enabling multiple run-starts daily and minimizing the need to coordinate schedules across end users. Flexibility is only meaningful if it simplifies your workflow or translates into cost or time savings.
At Element, we want to make it easier for scientists to explore their curiosity by putting them in control of their budget and their timelines. For more information on how AVITI can accelerate your single-cell research, watch our on demand webinar with 10x Genomics or fill in the form to talk to an Element scientist.
]]>urn:uuid:41765a07-80fc-44f8-8d41-5ba6f830a0e8In the race to develop new cancer interventions, time is money.2023-04-17T16:12:00-07:002023-05-19T08:34:18-07:00Felicia Hawthorne, PhDfelicia.hawthorne@elembio.com
The last thirty years have seen a 32% decrease in cancer death rates. The single largest driver of this success story is public health initiatives to reduce smoking, but improvements in early detection and treatment have also made significant contributions to better survival rates, particularly in breast and colon cancers. Next generation sequencing (NGS) has played a central role in these advances, first by transforming our understanding of cancer genomes, then as a critical tool for drug development and therapy selection.1 However, with cancer still the second most common cause of death worldwide, much more remains to be done.2 The desire to further improve outcomes is a driving motivation for cancer researchers, but success requires a clear-eyed view of what it takes to bring a new test or drug to market.
The Scylla and Charybdis of biopharma development
In 2021 alone, the biopharmaceutical industry invested over $200 billion in R&D, with the largest category being drug discovery and development in immune-oncology or other anticancer therapeutics.3,4 Bluntly, developing a new cancer intervention requires large financial investments and a tolerance for risk, and managing this risk often involves timelines. Every experimental delay lengthens program timelines, slows program reprioritization decisions, and increases the risk that funding will run out or that a drug or test will be too late to market to recoup investments. For biotech companies, both time and money are precious.
NGS sequencing is one area where this tension between time and money is clear. For cost efficiency, companies feel pressured to outsource sequencing to large, centralized labs to achieve low cost per base. However, this dependency on batching to achieve economies of scale introduces delays in turn-around times that slow research.
Breaking the link between scale and cost
Often when confronted with an either-or choice, the solution lies in thinking outside of the box – in this case, the DNA sequencing box. The AVITI™ DNA sequencing system offers biotech companies a way to save both time and money by breaking the link between scale and cost. The AVITI System is unique as a benchtop platform that combines high quality data, low cost per base, and maximum flexibility with 2 fully independent flow cells. At $2-5 per gigabase, running costs are on par with NovaSeq outsourcing costs, minus the time inefficiencies. The forthcoming Cloudbreak chemistry, coming in May 2023, will enable 2x75 runs to be complete in less than 24 hours. Best of all, the sample prep workflow is fully compatible with standard NGS library prep ecosystem partners, meaning negligible interruption to established lab processes.
An in-house AVITI system will enable you to:
Unlock resistance or prognostic markers of tumor evolution with somatic variant detection with up to 24 paired tumor/normal exomes per dual flow cell run.
Uncover tumor diversity and microenvironment roles in tumor progression by running exomes/genomes on one flow cell and transcriptome profiling on a second, either simultaneously or staggered.
Discover rare variants and novel biomarkers with deep targeted sequencing using validated solutions from Agilent, Twist, or Qiagen.
Sequence 10X Genomics single cell libraries at improved performance over SBS sequencing on fresh, fixed or FFPE samples.
Combine BD Rhapsody single cell library preparation, pairing cost-effective in-house sequencing with gentle handing of fragile and scarce immune cells and multianalyte detection.
Closing
In the race to score the next win again cancer, biotech companies are eager to find new ways to accelerate their research programs. If high sequencing costs or long project queues are holding back your innovation, an AVITI system can help you take control of your cancer research and development timeline.
]]>urn:uuid:eaaef781-ac19-4936-99b8-aab9ef874fb9AT AGBT Ag, agrigenomics scientists confront the challenges of food production in a changing climate head on2023-04-04T19:56:00-07:002023-05-19T08:36:32-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
If there is one takeaway from the 2023 Advances in Genome Biology and Technology – Agriculture (AGBT Ag) meeting, it’s that while the challenges of feeding the planet amidst climate change are many, the global mobilization to meet those challenges is equally impressive. AGBT Ag participants gathered in San Antonio, Texas to hear from an international array of speakers on wide-ranging topics, including how to extend the benefits of genomics to all, new strategies for connecting trait genotypes to phenotypes, novel approaches to genome editing, and newly available genomics tools.
AGBT Ag attendees had multiple opportunities to learn how agricultural researchers are already making use of the Element Biosciences AVITI™ System to accelerate their science. First, Carlos Congrains Castillo, a postdoctoral researcher at the University of Hawaii and the Scott Geib Lab at the USDA ARS, spoke at the Element Gold Sponsor Workshop about using the AVITI to improve the identification of invasive Tephritidae fruit flies. Carlos recounted that arthropods cause 20% of agricultural losses, and once established, are enormously expensive to eradicate. Early detection is key, but species with disparate ecological traits and economic impacts can be very hard to distinguish, especially in the larval and pupal stages that are typically found by fruit inspections at agriculture border checkpoints. One example is the mango fruit fly complex, comprised of 4 cryptic species. In this case, even DNA barcoding using standard sequences cannot adequately resolve members of the complex. Carlos discussed several strategies he is pursuing to improve the speed and accuracy of fruit fly identification.
Figure 1. The genome of A. fraterculus s. I was assembled from a sample shipped from Brazil at ambient temperature and sequenced with a combination of long reads and Hi-C data collected on the AVITI system.
One line of experiments involves combining long read sequencing with Hi-C data generated on the AVITI to generate reference assemblies from either lab grown or field-collected samples, where sample input and quality can be challenging. (Figure 1)
A second line of research involves developing better markers for genotyping. Carlos shared his work resequencing members of the mango fruit fly complex (the Bactrocera frauenfeldi complex) to identify markers in regions of high genomic differentiation to improve the speed and reliability of phylogenetic analysis. With high quality data generated on the AVITI, Carlos was able to pilot a new approach for rapid genotyping of B. frauenfeldi complex species by using pairwise FST to identify ~1,000 SNPs that are highly differentiated across the complex. Using only these markers instead of the full complement of 2.2 M SNPs provides near identical species resolution while significantly reducing the computational burden and analysis time.
Figure 2. Multiplexing 10 fruit fly samples on the AVITI instrument generated average coverage of 55x per sample, with highly consistent performance across five runs.
The next morning, early risers enjoyed Texas-style breakfast tacos in the Element lounge while taking in talks from three speakers. First, Meredith Ashby, Ph.D., Sr. Director of Marketing at Element gave an overview of the AVITI, the upcoming Cloudbreak™ chemistry release, and genomics applications for agricultural research. Next up, Adam Majot, Ph.D., Principal Scientist at Agriplex Genomics, shared early results of onsite Cloudbreak beta testing. Agriplex uses the AVITI to provide GBS services to customers. For the beta test, they compared Cloubreak and V1 genotyping performance on 3,096 rice leaf crude extracts labelled with 15 bp UDI barcodes. Highlights of his presentation included ongoing high accuracy between v1 and Cloudbreak chemistry, 99.86% concordance between genotyping calls, and impressively consistent index representation across chemistries, as compared to Illumina same-chemistry replicate runs.
Figure 3. Ultra-lowpass sequencing on the AVITI with coverage as low as 0.1x generates genotyping calls concordant with a higher coverage truth set. Libraires were prepared with the seqWell plexWell Low Pass kit and data was analyzed with the Gencove platform.
Finally, Jesse Hoff, Ph.D., Agrigenomics Product Manager at Gencove, rounded out the morning with a presentation of his poster, “Highly accurate, cost-effective genotyping with ultra-lowpass whole genome sequencing.” Jesse described the results of multiplexing 1,536 Angus and Holstein bovine samples using seqWell plexWell Low Pass 384 kit, sequencing with a 2-flow cell run on the AVITI, and imputation and genotyping with the Gencove analysis platform. The study found that coverage as low as 0.1x provides 99% accurate genotypes compared with a higher coverage truth set, demonstrating that ultra-lowpass on the AVITI is still sensitive enough for genomic selection while providing sequencing costs as low as $1.50 per sample, depending on annual volume.
With thought provoking conversations, wide-ranging scientific talks, and a farewell banquet complete with cowboy hats and line dancing, we are already looking forward to AGTB Ag 2024!
]]>urn:uuid:ff9fd48b-42a5-40d4-b88b-6fb97351c1e1Low pass whole genome sequencing 101: AgBio and the future of genotyping2023-02-22T18:23:00-08:002023-12-01T10:10:02-08:00Meredith Ashby, PhDmeredith.ashby@elembio.com
With the steadily decreasing cost of sequencing, low pass whole genome sequencing with imputation is gaining traction in agriculture as a method of genotyping. In this Element 101 blog series post, we dig into how low pass WGS works, what it can offer beyond targeted genotyping options like PCR and microarrays, and how the Element AVITI™ system can help even moderate throughput labs make the leap to this high information, cost-effective method.
Balancing information density and cost per sample
In an ideal world, genotyping could be done using 30-fold coverage whole genome sequencing, supplying high confidence data across all possible variants in one easily operationalized assay. However, the high cost per sample combined with the slow turnaround time for whole genome sequencing at the scale required for commercial genotyping makes this impractical. Microarrays have long been an appealing solution for genotyping, as they offer:
Low cost
Ease of use
Scalable number of markers
However, microarrays come with their own compromises:
Requirement for a reference genome
High design, startup, and revision costs
Low potential for variant discovery, as only known targets can be included on microarray chips
Potentially poor sampling of genetic diversity from non-domesticated or divergent strains
As the cost of genome sequencing has continued to drop, new targeted sequencing-based methods have been developed as ways to improve the discovery power of genotyping methods while still controlling cost per sample. One popular method of genotyping by sequencing (GBS) involves DNA digestion with restriction enzymes, followed by adapter ligation, amplification, and sequencing. Another variation involves highly multiplexed PCR reactions to amplify known markers, followed by amplicon sequencing. Hybridization capture is yet another alternative for focusing sequencing on markers of interest. All methods offer notably improved potential for the discovery of new variants linked to important traits. GBS methods come with their own limitations, though:
Like microarrays, GBS requires a reference genome and significant investments in assay development and validation.
The manufacture of probes for target capture GBS requires a large initial investment and economies of scale to achieve favorable pricing.
The target capture protocol is laborious and requires relatively high DNA input quantities.
Working with GBS service providers can make these methods more accessible, particularly for commonly studied organisms, but outsourcing can increase turnaround times and it may be preferable to maintain internal control of samples.
Low pass genome sequencing paired with imputation has the potential to overcome many of these remaining barriers to the adoption of NGS for genotyping by combining assay simplicity, high information content, and affordable cost per sample. In this method, samples are sequenced to a depth of just 0.4 to 1.0-fold coverage and imputation analysis is employed to backfill missing sequences using prior knowledge of gene variant co-inheritance patterns. While a reference genome is still needed for genotype calling from low pass sequencing data, the advantages over microarrays, in particular, are clear:
No reagent startup costs related to assay development or redesign.
Operational simplicity, with commercially available options for high throughput library preparation and data analysis support.
Markers across the entire genome can be leveraged for simultaneous trait selection and variant discovery.
As more samples are sequenced and added to a low-pass WGS database, the potential for discovery of novel variants increases, including through re-analysis of archived data.
Fig 1: Low pass sequencing plus imputation captures most of the benefits of whole genome sequencing for genotyping but at dramatically lower cost.
Low pass WGS on the Element AVITI system
Low pass genome sequencing has the strong potential to simply and accelerate breeding programs, however it is only now becoming practical from a cost standpoint. In order to achieve favorable pricing, users must typically be able to batch very large numbers of samples for sequencing on ultra-high throughput systems. This has been achievable for large breeding operations where samples can be run centrally, or by smaller operations that are able to outsource. Now, however, the Element AVITI DNA sequencing platform offers an alternative.
The AVITI is unique as a highly accurate, mid-throughput DNA sequencer with cost per gigabase comparable to production scale systems. At ~$5 per gigabase, low pass sequencing is affordable even with dramatically fewer samples to run. The AVITI also offers technical advantages, including negligible index hopping and high effective coverage stemming from a low duplication rate.2
Customers with mid-level sample throughput can affordably run samples in-house, reducing turnaround times while maintaining possession of valuable materials and data. For large-scale operations, the AVITI opens the door to decentralization. Turnaround times can be reduced by having genotyping facilities at or near globally dispersed production sites to take advantage of year-round growing cycles. With throughput based pricing available through Element’s $200 Genome Program, sequencing costs can be further reduced to as low as $2 per gigabase.
Fig 2. At 20,000 samples per year assuming a 2.4 Gb genome and 0.5x coverage per sample, the AVITI achieves lower costs than the NovaSeq using an S2 flow cell. Calculations assume all flow cells are maximally utilized to achieve lowest sequencing cost per sample. Comparison does not include preparation, analysis, and warranty costs.Fig 3. At 100,000 samples per year, the throughput-based $200 Genome Program allows comparable pricing to the NovaSeq on an S4 flow cell, with the added potential benefit of decentralization. Calculations assume all flow cells are loaded in multiples of 96, for enhanced operational efficiency. Comparison does not include preparation, analysis, and warranty costs.
While switching over to a new genotyping method is a significant lift, the AVITI presents a new route for breeding operations of all scales to affordably increase the power of their genotyping pipeline. The implementation of a new workflow can be simplified by working with validated Element ecosystem partners for both library prep and imputation analysis. Multiple options exist for high-throughput library preparation for the AVITI, including purePlex from seqWell, a Tn5 transposase-based system that facilitates even pooling of libraries at scale. For analysis, Gencove
offers an enterprise analytics platform for low pass imputation that simplifies genotype calling and report generation.
To learn more about whether the AVITI is right for your lab, talk to an Element scientist by filling in the form on this page, or contact us here.
References
1. Li et. al. (2021). Low-pass sequencing increases the power of GWAS and decreases measurement error of polygenic risk scores compared to genotyping arrays. Genome Res. 31(4):529-537. doi: 10.1101/gr.266486.120.
2. Li et. al. (2022). Low-pass sequencing plus imputation using avidite sequencing displays comparable imputation accuracy to sequencing by synthesis while reducing duplicates. BioRxiv. doi: 10.1101/2022.12.07.519512.
]]>urn:uuid:34cc9759-804a-4b65-a7af-1c0a345dcff8Element Biosciences showcases new science and upcoming chemistry advances at AGBT 20232023-02-15T08:46:00-08:002023-05-19T09:20:30-07:00Meredith Ashby, PhDmeredith.ashby@elembio.comMatt Kellinger, PhD - VP Biochemistry and Co-Founder
Element came out in force last week at the annual Advances in Genome Biology and Technology (AGBT) conference in Hollywood, FL, with an array of lounge talks and program appearances, as well as a workshop announcing details and data supporting the upcoming Cloudbreak chemistry release and 2x300 cycle kit releases.
The week started out with three application-focused talks in the Element hospitality lounge. Carrie Cibulskis, Director of Cancer Genomics at the Broad's Genomics Platform, started the presentations with her evaluation of AVITI™ for cancer sequencing applications commonly used by Broad researchers. Cibulskis noted that the AVITI already shows matched performance to Illumina sequencing across a range of cancer genomics applications, including shallow depth, exome, and panel sequencing, even without platform-specific analysis pipeline optimization. “I didn’t expect it to look so good on the first try, to be honest,” she said. “We are really encouraged by this data." For more details, review her team’s poster or watch the video of her presentation.
Figure 1. Carrie Cibulskis shared that Element AVITI data performs comparably to Illumina data across a range of cancer sequencing applications, including exome, low pass, and targeted panel sequencing.
Next, Jon Armstrong, VP of R&D at Jumpcode Genomics, presented work showing the utility of CRISPRClean for reducing noise and uncovering hidden signals in single cell RNA sequencing data on the AVITI.
Finally, Element VP of Informatics, Semyon Kruglyak, presented a deep dive into the high quality scores of AVITI data and the unique capabilities of Element technology, illustrating how these translate into meaningful advantages for customers. He shared new data showing that AVITI read quality is particularly higher than NovaSeq data in post-homopolymer regions, with potential impacts to variant calling. He also presented internal research on sequencing library inserts greater than 500 bases, enabled by our method of polony generation using rolling circle amplification. Modelled perfect data shows that paired end sequencing of longer inserts provides many of the same advantages as continuous long reads for improved mapping and variant calling. Semyon showed that by sequencing libraries up to 2 kb in length, the AVITI system can provide sufficient coverage in hard to sequence regions of the human genome to enable SNP calling.
Figure 2. Sequencing libraries with 500 bp inserts (top panel, BBS-0174) can result in coverage gaps in repetitive regions where there is no or low-quality mapping. In contrast, sequencing libraries with median insert size of 1 kb (bottom panel, APP-0680) provides enough coverage for variant calling in regions that have previously been less accessible to short reads.
Another highlight of the conference was a presentation by Elinor Karlsson, Director of the Vertebrate Genomics Group at the Broad, professor at the UMass Medical School and the “dog whisperer” of genomics. She presented her research on the genomics of dog behavior from the main stage, noting that she is now sequencing on AVITI with good results. The heterozygous variant calling "is as good as with Illumina, and possibly better," she told GenomeWeb.
On Day 2, Element hosted a second series of presentations in our lounge. First, Francisco Garcia, SVP of Software and Informatics, presented a preview of AVITI OS 2.0 and Elembio Cloud. This new version of the AVITI OS includes a simplified user interface, automated data streaming to your desired data location, and adds onboard demultiplexing that provides early feedback on pool balancing. Elembio Cloud delivers the ability to set up and monitor runs remotely, and consolidated options for data storage and management.
Next up was a scientific open house, with Chief Scientific Officer and SVP, Applications, Shawn Levy joining Co-Founders VP of Chemistry, Matt Kellinger and Chief Technology Officer, Mike Previte taking questions from the audience on a wide range of topics, including what makes Element technology unique and how our scientific leadership plans to turn those key differentiators into concrete benefits for the genomics community in the coming months and years.
Finally, Matt Kellinger again took the main stage for a bronze workshop presentation on "Cloudbreak and Beyond." He shared improvements our new chemistry is bringing to AVITI less than a year into launch. The smart phone cameras rose excitedly when Matt started showing the difference between AVITI and NextSeq in performance, with AVITI delivering huge cost savings, better accuracy, and runtimes faster by 10 hours. In addition to the advances shared in our lounge talks, Matt highlighted Cloudbreak improvements including:
Even higher accuracy, especially in read 2
Faster run times, with 2x150 runs completing in 38 hours
On-board circularization of AVITI-native Elevate libraries, with no increase to run times
A preview of 2x300 runs delivering 400 M paired end reads per flow cell, coming in 2023 Q3
Figure 3. Matt Kellinger showed a preview of Element’s forthcoming 2x300 cycle kit, coming this fall.
Thanks so much to everyone who stopped by our lounge to see an AVITI demo, came to our lounge talks and workshops and joined us for some late-night fun at our 'Catch the New Wave’ party. If you weren’t able to attend, we hope you enjoy the recordings of all our AGBT happenings and check our events page to see where to find us at future events and webinars!
]]>urn:uuid:19be1f0b-b4f4-4d9d-9960-cc5427329c8eTaking Bold Steps into the Future to Redefine What is Possible2023-02-08T13:47:00-08:002023-07-01T20:02:39-07:00Molly He, PhDmarisabatey@gmail.com
By Molly He, CEO & Co-Founder, Element Biosciences
When faced with uncertainty, sometimes it pays to be bold. The bold among us can see opportunities. At Element we are strategic risk-takers who see opportunities for innovation everywhere. We are not afraid to disrupt the norm.
At the J.P. Morgan Healthcare Conference last month, we announced the availability of a $200 genome on our AVITI™ Benchtop Sequencing System. We wanted to provide a flexible, volume-based program for customers to get access to the highest quality sequencing at the lowest cost. With AVITI we can offer that $2/G without requiring customers to batch samples or sequence 20,000 genomes a year. While some companies have talked about driving down costs for customers, I am glad to say that we have delivered on that promise today.
Looking back on what we aimed to build in 2018, we can see that Element has achieved what we set out to create and we are uniquely positioned to disrupt the industry with an unmatched combination of performance, cost, and flexibility.
Less than a year after the launch of AVITI, we are producing the best data quality on the market. AVITI has fewer optical duplicates, less index hopping, and significantly fewer errors in sequences that follow homopolymers compared to Illumina. Data from our customers and partners show the strength of our technology. For example, looking at exome data from Qiagen using the same library run on NextSeq550 and AVITI, the vast majority of our reads on AVITI are >Q42. For whole genome sequencing, researchers at UC San Diego demonstrated that AVITI’s higher Q scores translated into good clinical yield, which is very important in the clinical setting.
AVITI is superior for high-quality, low cost exome
We are also beating the competition on price. Running medium throughput of three flow cells per week on AVITI saves customers about $1.7 million over three years vs. NextSeq 2000 at list prices. Imagine how much more science can be done! We have seen some drastic discounting by Illumina. Even if they gave away a NextSeq 2000 for free, sequencing on the AVITI would still be more cost-effective, allowing you to break even in under six months.
This year we have an exciting outlook ahead as we introduce our Cloudbreak chemistry in Q2, bringing 20 percent faster run times and simplified workflow, and in Q3 our new 2x300 kit, which has a factory-scale price per G comparable to the NovaSeq. We will be presenting more details at the upcoming 2023 AGBT meeting.
AVITI is redefining what is possible on a desktop, and we are just getting started. Working together with our talented and dedicated team at Element, our customers and partners, I am confident we will transform AVITI into the most versatile omics tool ever created. We will remain steadfast in our unyielding commitment to increasing access to genomics everywhere and catalyzing breakthrough science.
]]>urn:uuid:e3d8afe6-502e-42c3-adcb-afb64c2d57baThe AVITI™ System for 10X Genomics single cell analysis2023-02-08T13:19:00-08:002024-08-07T13:02:43-07:00Isaku Tanida, PhDisaku.tanida@elembio.comThe AVITI™ System for 10X Genomics single cell analysis
The AVITI™ System for 10X Genomics single cell analysis.
Unmatched savings and throughput flexibility
(transcript)
Hi, I'm Isaku Tanida with Element Biosciences and today I'm going to go through a review of how the AVITI system combines with 10X Genomics technologies to create a powerful solution for single cell analysis. In particular, I'll cover how the AVITI flow cell and instrument design lines up well with the configuration of the Chromium chip and of course, the enormous sequencing costs and time savings that you'll achieve through the upcoming upgrades to our avidite chemistry.
Hopefully you've already seen the recent announcement of Cloudbreak, an upcoming enhancement to our sequencing chemistry that enables it to significantly reduce sequencing times.
With Cloudbreak, typical single cell assays require less than 24 hours to run, combined with its lowest cost per read on a benchtop instrument AVITI is truly setting the standard for single cell sequencing. So over the next few slides, I'll show how running the AVITI with Cloudbreak chemistry would allow you to increase the frequency of experiments you complete per week, and that would generate an astounding level of savings in sequencing costs compared to SBS alternatives on an annual basis.
To quickly view in this figure. You see the 10X single cell workflow at a very high level when combined with the AVITI system to prepare for sequencing.
10X single cell libraries are generated as they normally would be, and resulting libraries are then circular, using a simple process at the bench for what's called the Adept Library workflow. You can easily understand the general idea that each of these steps are completed using kitted solutions.
But the next two slides, I'm going to first describe the Adept process in a little more detail so you can understand how it prepares a 10X single cell library for sequencing. And second, I'll describe how the configuration of the 10X Chromium chip aligns well with the very system such that little is wasted or time delayed by moving from library preparation to sequencing.
This figure shows the entire molecular workflow so you can visualize what and why each of the steps are necessary and how they happen. In the left panel, each set of the wells in the 10x chip is loaded with cells and barcoded beads to create gel beads in emulsion, with up to eight samples per chip, each sample well has thousands of cells. What follows is all standard 10X genomics processes that result in the expected linear DNA library pictured on the top right panel. These libraries all contain the critical P5 and P7 sequences that are needed as inputs for the Adept library compatibility workflow pictured beneath it.
The Adept workflow is a circularization process that requires no PCR amplification to achieve, which avoids the introduction of potential bias or error through this step. In this slide, you see a mocked up schedule of sequencing runs where single cell assays are run each of the first two days at 9 a.m. due to the 24 hour runtimes that can be achieved on DVD. For assays running up to 150 cycles. In this example on day one at 9 a.m. indicated as d1-9a, the schedule starts the equivalent of two sets of eight chromium samples on flow A and flow B simultaneously.
Each with different cell numbers per sample, a different read depth per sample set. On day two beneath it on the schedule, a single set of eight samples from a Chromium chip is split across the two flow cells. A and B, again, each flow cell running different cell numbers and read depths
On day three at 9 a.m, the schedule has an exome study on side A, but then in the middle of that exome run on the same day, a new round of single cell sequencing kicks off at 4 p.m. without disrupting the exome run on side A
In summary, unlike other benchtop space systems, the AVITI operates two independent flow cells versus one. And unlike other benchtop platforms, with multiple flow cells, the AVITI does not need the system to complete a flow cell run, before beginning another one.
Lastly, let's take a look at flexibility, throughput and cost versus alternative choices over the course of a year. In the previous slide, you can see how to generate large amounts of data daily on the AVITI. That frequency allows you to iterate on your experiments more often.
And you can see how doing this doesn't come at a penalty. In fact, on the average, you enjoy a tremendous cost savings compared to any of the other choices listed on this table. In this table, running four flow cells per week was used as a model. At that rate compared to the NextSeq 2000 this would generate over $500,000 in savings each year by comparison. The NextSeq2000 is also greatly hampered by the fact that it only operates a single flow cell which provides no flexibility per run. By comparison, the NovaSeq SP flow cell offers a faster runtime and the dual flow cell option but with zero savings compared to the NextSeq. You pay a premium for that added incremental level of speed and flexibility because you captured no savings improvement over the NextSeq. Skipping ahead to the S2 flow cell with its larger batch size, the S2 offers a 50% discount in cost compared to the SP, but it still falls behind the AVITI by $280,000 per year with a burdensome 4X batch size requirement. Even the S4 flow cell on the far right does not offer a break even costs versus the AVITI on an annual basis and requires you to wait for a full two week equivalent of sample batching.
From this table you can see the AVITI uniquely allows you to both pilot smaller batch size experiments on a daily basis at great cost savings while you also while also allowing you to scale output substantially by combining dual flow cell runs daily.
Another way to put it - you can generate 10 billion reads or the equivalent of an S4 flow cell by running five pairs of 24 hour runs on in the AVITI, while always maintaining the flexibility of what samples you run on each flow cell each day.
I hope this review was helpful to more clearly understand how the system can plug seamlessly into your ten single cell experiments, while offer you a cost savings and flexibility that other leading systems simply cannot match. Thank you again to our partners at 10X Genomics for their ongoing collaboration and support.
For more details on the performance of DVD with the 10X platform, please feel free to download our 10X datasets from the Element Biosciences website and look for upcoming news where we combine the AVITI system with 10X single cell assays for fixed tissues.
Thank you.
]]>urn:uuid:5221bac0-4ed5-460d-9c61-60a264d00375At PAG 2023, Element introduces the AVITI™ system to the AgBio research community2023-01-31T10:41:00-08:002023-05-19T08:46:59-07:00Meredith Ashby, PhDmeredith.ashby@elembio.comElement Biosciences: Low Pass Sequencing in Animals
What better way to start the year than attending our first PAG, the largest international gathering of plant and animal genomics scientists in the world, held right in our backyard in San Diego, California. We were excited to see just how many attendees came eager to talk to us about Element’s mission to disrupt genomics with high-quality, low-cost DNA sequencing data on the AVITI. Coming on the heels of our $200 Genome Program announcement at the JP Morgan Healthcare conference, PAG participants were ready to learn how they, too, could benefit from our new throughput-based pricing model to further bring down the cost of sequencing for the applications most important to them, including whole genome sequencing, genotyping by sequencing, low-pass genome sequencing for genotype selection, and RNA sequencing.
Figure 1. PAG attendees came to the Element booth to see for themselves how the AVITI system works.
In addition to instrument demos, PAG attendees had multiple options for seeing AVITI system data on the scientific program. On Monday, Alison Devault from Daicel Arbor Biosciences presented a poster “A universal targeted sequencing system for any high-throughput sequencing platform,” where she described the myBaits® enrichment system, which is compatible with virtually any input nucleic acid and sequencing platform. The poster featured data showing that for five distinct assay types, the AVITI returned comparable or better library representation, reads on-target, and locus retrieval as NovaSeq despite highly variable length distributions and target compositions."
That same afternoon, Jacky Carnahan from Neogen Genomics presented a poster “Low pass sequencing in animals: Element Biosciences AVITI as a useful platform.” The poster featured a three-way collaboration between Neogen, Gencove and Element, describing an end-to-end solution for low pass sequencing and genotype imputation. The genotyping calls were highly consistent with results from a production-scale DNA sequencing instrument, but lower duplication rates yielded higher effective coverage. Additionally, the reagent cost per Gb was comparable to the higher throughput system, providing a cost-competitive path to potentially faster turnaround times at lower sample volumes.
Finally, Beth Rowan presented preliminary data on her methods development project, “Evaluation of Pooled and Single-Nucleus Sequencing Approaches for the Study of Recombinant Populations and the Generation of Genetic Maps.” The overall goal of her study is to use CRISPR to alter genes involved in the regulation of meiotic recombination and for that she needed to develop a rapid method for detecting the frequency and locations of meiotic crossovers. Pooling and sequencing recombinants offers an efficient, high-throughput readout for assessing changes in crossing-over events after genome editing, with TELL-seq linked read sequencing and single cell RNA sequencing as two options for finding crossover breakpoints in recombinant populations. Dr. Rowan reported that the TELL-seq method looks both promising and cost effective in a low coverage test run, and she plans to scale up her pilot, potentially on the AVITI. She also shared preliminary data on single cell RNA-seq using Parse and sequencing on a shared AVITI flow cell. While this approach will require further optimization to reduce rRNA representation in the data, Dr. Rowan noted that “The AVITI data was high quality, and I can recommend the system as a practical and cost-effective option for methods development pilot studies.”
Figure 2. Beth Rowan outlines her initial workflow for using single cell sequencing as a readout for meiosis perturbation experiments.
]]>urn:uuid:1b657f71-51ee-4dfd-8f88-ec0db9d286c3Somatic and Germline sequencing of SureSelect Libraries on the Element AVITI System2023-01-25T13:28:00-08:002023-05-19T08:48:29-07:00Felicia Hawthorne, PhDfelicia.hawthorne@elembio.comFelicia Hawthorne, PhD Explains Somatic and Germline sequencing of SureSelect Libraries on the Element AVITI System
Summary Transcript
This is a brief overview of our experience sequencing germline and somatic samples with Agilent's SureSelect Library Prep on our AVITI system.
This study was done on a pre-release early version of AVITI, and the results of the study were made available as a data sheet by Agilent. SureSelect library prep kits were used for both somatic tumor and germline normal samples.
For the normal samples, the all human exon panel was used and a custom cancer panel was used for tumor samples. This collaborative study demonstrated the ability to leverage the current Agilent SureSelect catalog and their custom workflows without the need to develop new library prep solutions or protocols for the AVITI system.
The AVITI sequencing system can run independent flow cells on either side of it. So on one side, you could run your germline exome samples while the other runs your somatic targeted samples. Using the 2X150 sequencing kit each flow cell can generate up to a billion reads in under 48 hours. So you can run 60 paired germline and somatic samples and yield 5G per sample. To assess the sequencing quality we looked at the percent passing filter and Q30.
And as you can see in the table with the germline exome panels and the targeted panels, deeper sequencing of somatic samples yielded high percent passing filter and high Q30. Now again, remember, this was done using a prerelease version of the product.
So you'll find the data published from studies on the launch version of the product have even better data quality. Next variant calling performance for germline samples shown in the top table were evaluated by looking at the recall or percent known variants called correctly and precision or percent of calls that were known variants.
While for somatic samples shown in the bottom table, we assessed their accuracy of detected versus expected frequency for variants. And as you can see, a high alignment was demonstrated even for variants with 5% and lower frequency. Additionally, key sequencing metrics such as fold 80, duplication rate, and coverage demonstrated robust sequencing data generated from germline exome samples and deeper somatic samples where higher duplication rates are expected.
I hope this overview is helpful to highlight how easily the AVITI system can enable you to do more as you plan your next project. I would also like to extend our thanks to our partners at Agilent for their collaboration and support.
For more details on the performance of the AVITI with SureSelect Library Prep for germline and somatic samples, please feel free to download the datasheet from the Element website and look for new products using the AVITI system. Thank you.
]]>urn:uuid:fbbca2ac-272e-40a8-b9f1-b36f97547c64A New, Less Expensive Path to Methylation Detection in place of Bisulfite Sequencing2022-11-30T15:29:00-08:002024-04-19T16:40:00-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
Cytosine methylation plays an important role in many biological processes, including cell lineage specification, X-chromosome inactivation, and the preservation of chromosome stability. Given its importance in so many basic cellular processes, errors in methylation have been linked to a wide range of human diseases, including cancer and autoimmune disease.
The detection of methylation patterns via DNA sequencing is an important tool for researchers trying to unwind the mechanisms of human disease and health, but it these experiments can be expensive due to the inherent low diversity of bisulfite samples and the limitations of traditional NGS technology.
The Challenges of Bisulfite Sequencing
To assess the methylation state of DNA via sequencing, it is common to convert unmethylated C’s to U’s either enzymatically or via bisulfite conversion. Following sequencing and an alignment to both a methylated and unmethylated reference, the methylated sites can be identified in the sample of interest.
However, because the vast majority of cytosines are unmethylated in most sample types, this leads to very few C base calls (and an overabundance of T base calls) in the resulting sequencing library. Libraries with one or more under-represented bases are termed low diversity and they pose a challenge for many sequencing technologies.
With many technologies, low diversity samples interfere with the ability to map the location of distinct clusters and maintain base calling accuracy as sequencing progresses. To overcome this challenge, it is common to either pool such libraries with high diversity libraries or to supplement the library with a significant PhiX DNA spike-in prior to sequencing. While amount of PhiX DNA required and the impact of low diversity on target density varies by sequencing platform, the addition of reduces the effective throughput of the run, driving up the cost of sequencing.
A Better Path to Low-Diversity Sequencing on the AVITI™ System
Unlike other sequencing platforms, the AVITI system does not require diversity to maintain accuracy as signals from the four bases are more reliably distinguishable due to our unique sequencing chemistry. In addition, AVITI libraries have specific characteristics that enable clean mapping of polonies during the initial cycles.
We decided to assess the capability of the AVITI Sequencing system on MethylSeq libraries. Our objective was to evaluate density and accuracy, while varying the PhiX spike-in percentage. Libraries were prepared using the NEBNext Enzymatic MethylSeq kits. Figure 1 shows the library preparation process.
Figure 1: Library Preparation Process
We sequenced the well-characterized sample NA12878 pooled with the addition of 1% each of a fully methylated control library (pUC19) and a fully unmethylated control library (phage lambda). Three runs were completed with a PhiX spike-in of 0%, 5%, and 20%, respectively by concentration. The summary of the primary sequencing metrics is provided in Table 1.
Condition
PE Reads
%Q30
PhiX Aligned (%)
No PhiX
857 M
94%
N/A
5% PhiX
920 M
95%
5.3%
20% PhiX
989 M
96%
27%
Each run surpassed the 800M PE specification and attained a high percentage of Q30 bases. The PhiX spike-in for the third condition was higher than expected, reflecting either loading variation or some amplification preference for the PhiX reads.
We processed the run using the NFCore implementation of the Bismark methylation pipeline to obtain the percentage of methylated CpG sites in each of the libraries. According to documentation from sample manufacturer NEB, the expected methylation percentage of CpG sites for the 3 libraries are 53% (NA112878), 100% (pUC19), and 0% (phage lambda). Figure 2 shows our results from the output of the Bismark pipeline.
Figure 2. The three panels show the percent methylation of NA12878 (A), puc19, (B) and PhiX (C) under each of three different PhiX spike-in levels.
Avoid the PhiX Tax to Lower Your Sequencing Cost
The methylation fraction closely matches the expected results and is highly consistent across runs even with very little PhiX present. The FASTQ data is publicly available on our website. These results show that the AVITI system is compatible with the NEBNext Enzymatic MethylSeq and produces high quality methylation data, even with no PhiX spike-in.
We still recommend a 5% PhiX spike-in for robustness and real-time error measurement.
However, a large amount of PhiX is not required to obtain accurate MethylSeq data on AVITI, further lowering the cost of sequencing relative to a competing mid-throughput platform by a further 15%, on top of the already lower cost of reagents.
]]>urn:uuid:4ccc37df-f9b8-4ec8-a282-44bcb85d96bfContext Matters: Isoform Sequencing Reveals a Higher Resolution Picture of Colon Cancer Progression2022-11-08T10:00:00-08:002023-05-19T08:57:51-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
In a recent paper published in Nature Communications Biology, researchers showed that synthetic long-read sequencing using Element Biosciences’ LoopSeq™ technology identified changes in isoform expression that enhanced researchers’ ability to distinguish among cancerous, metastatic, and normal tissues in a study of colon cancer samples. Further inquiry into isoform-specific changes in gene expression may reveal new biomarkers, drug targets, and insights into cancer progression.
LoopSeq provides highly accurate, long-read information on short-read sequencers without needing a dedicated instrument. When used for RNA sequencing, LoopSeq provides full-length isoform data that short reads cannot. This information is particularly valuable for understanding changes in gene expression in cancer, where structural variants, mutation phasing, gene fusions, and aberrant splicing are known to impact disease progression, severity, or treatment response. Better information about this molecular “dark matter” can reveal novel therapeutic targets or serve as biomarkers to improve early detection.
LoopSeq: A Validated Alternative to Native Long Reads
The Nature Communications Biology study was led by Jianhua Luo, MD, PhD, Professor of Pathology at the University of Pittsburgh, and Tuval Ben-Yehezkel, Senior Director of Applications at Element Biosciences.
Luo, Ben-Yehezkel, and colleagues first validated the LoopSeq method by sequencing Hela total RNA spiked with the External RNA Controls Consortium (ERCC) sample and comparing the data to previously published results from long-read sequencers. LoopSeq produced a .01% per base error rate, which was lower than available results from PacBio, Oxford Nanopore, and Illumina sequencers.
Isoforms as Markers of Cancer Progression
The researchers then used LoopSeq to study human colon cancers, sequencing control tissue, primary tumors, and lymph node metastases. The researchers used probe-capture oligos to capture the split regions of the 2,193 most common cancer-related gene fusions found in the TCGA database. With LoopSeq data, the researchers could quantitate differentially expressed genes (DEGs) and differentially expressed isoforms (DEIs) across the three sample types. Remarkably, they found that while hierarchical clustering based on DEGs did not adequately distinguish between tumor and metastatic samples, clusters that instead relied on DEIs did. (Figure 1)
Figure 1. DEIs provide better resolution of the normal, tumor, and metastatic samples than DEGs.Figure 2. Changes in isoform distribution correlate more closely with the cancer stage than changes in gene expression alone.
Drilling further into what aspect of gene expression data provides the most information on cancer stage, the authors considered DEGs with unchanged isoform patterns, DEGs with changes in isoform distribution, and DEIs with no net change in gene expression level.
Interestingly, focusing on isoform redistribution without gene expression changes produced the best tissue-differentiation results (Figure 2).
The authors note that “DEIs, which might have previously been inaccessible and were hidden within comparable gene expression levels, represent an additional dimension in differential expression analysis.”
Another notable finding was the detection of single nucleotide variations (SNVs) that were unique to specific isoforms. The researchers found 4,042 SNVs in the six cancer samples. Of the 1,509 SNVs found with at least two isoforms and five assembled contigs, 86% were not distributed evenly.
Finally, four previously unknown fusion genes were found in the LoopSeq data using SQANTI, a bioinformatics pipeline for classifying long reads by splice junctions. One novel fusion, STAMBPL1-FAS, was initially discovered in just two of the metastasis data sets but was later found in all of the study cancer samples using qPCR.
STAMBPL1 is a deubiquitinase inhibitor of apoptosis in the nNF-kB signaling pathway, whereas FAS is a cell surface death receptor.
Isoform Context Expands Our View of Cancer Biology
While quantitative RNA sequencing has long been used to understand the impact of genetic mutations on tumor biology, this proof-of-concept study demonstrates how layering in isoform information can add richness to these analyses. Isoform data can reveal changes in gene expression that are invisible to short-read methods but correlate strongly with the tumor stage. In addition, long-read sequencing continues to reveal new gene fusions, some of which may have wide distribution. LoopSeq provides a new, more accessible option for generating long-read data that can further our understanding of cancer biology.
Contact us to purchase our LoopSeq 16S or Amplicon Sequencing Kits or get a quote for LoopSeq service.
Source for figures: https://www.nature.com/articles/s42003-021-02024-1
Figures 1 and 2 for the blog are Figure 2A and Figure 2C from the paper, respectively.
]]>urn:uuid:031e56a4-c419-4c71-964d-9243ef999018LoopSeq™ Kits for the AVITI™ System: Democratizing Access to Long-Read Sequencing2022-11-02T13:17:00-07:002023-05-19T09:01:05-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
Long-read DNA sequencing has gained broad acceptance as an important tool for genomics research, providing biological insights in a wide range of contexts where short reads can only reveal part of the picture.
However, significant barriers still need to be overcome for scientists leveraging this method to advance their research, including cost and timely access to dedicated long-read instruments. Highly accurate long-read sequencing, in particular, has a very high cost per Gb, and sample queues can be months long.
To help scientists better access this technology, we are launching new LoopSeq long-read sequencing kits designed for the AVITI platform and other short-read platforms.
Expanding the AVITI Application Menu With Long-Read Sequencing
Element LoopSeq is a synthetic long-read technology that turns your short-read system into a long-read platform. It provides complete coverage of long DNA molecules, generating a data type identical to the output of legacy long-read technologies such as PacBio and Oxford Nanopore but without needing an additional dedicated instrument. Watch our short technology overview video to learn more about how LoopSeq works. With the release of our new LoopSeq for AVITI kits, it has never been easier or more affordable to access long-read sequencing.
The New LoopSeq Kits Come In Two Configurations:
For microbiome researchers, 16S LoopSeq for AVITI contains all reagents required to amplify full-length 16S, complete the LoopSeq workflow, and generate one multiplexed library of up to 96 samples for sequencing on the AVITI system.
Amplicon LoopSeq for AVITI enables the same 96-sample workflow but for any amplicon of choice. For researchers who don’t need to process 96 samples at once, Extension LoopSeq for AVITI allows users to make additional sequencing libraries from unused 16S LoopSeq for AVITI or Amplicon LoopSeq for AVITI reagents.
For non-AVITI owners, 16 LoopSeq, Amplicon LoopSeq, and Extension LoopSeq are platform-agnostic kits.
Genomic Context Matters
While short-read sequencing is the workhorse of genomics, there are many targeted sequencing applications where additional genomic context can change or add nuance to our understanding of biological systems.
The true diversity of bacterial communities can only be adequately resolved with full-length 16S sequencing.
Novel isoforms, or changes in relative isoform abundance can only be discovered with reads that span all exon junctions.
Full-length immune repertoire sequencing provides access to CDR3 regions that cannot be accurately assembled with short reads.
The evolution of viral quasispecies in response to a host immune response can only be fully mapped by phasing variants with long amplicon sequencing.
In all these cases, the ability to phase complete genes can reshape our understanding of biological systems rich in diversity. Our LoopSeq for AVITI kits make it easier than ever for researchers to accelerate their science with high-quality long-read data.
]]>urn:uuid:256850c6-5fcc-4fb4-88fa-a63d2ee88759Count On Element Biosciences to Make Single-Cell RNA Sequencing More Affordable2022-10-25T12:29:00-07:002023-05-19T09:23:52-07:00Meredith Ashby, PhDmeredith.ashby@elembio.com
Single-cell RNA sequencing has rapidly become a critical tool for scientists who study tissues where transcriptional diversity drives biological functions, including immunology, neuroscience, cancer, stem cell research, and developmental biology. Single-cell sequencing is a relatively new tool with many open challenges, including dropouts and the need for new data science methods, but managing the cost of experiments is almost always a significant consideration.
Here, the AVITI™ System represents a new, disruptive force in next-generation sequencing (NGS) technology that offers exceptional DNA sequencing accuracy at a fraction of the typical reagent cost. By leveraging low-binding flow cell surface chemistry, polony generation with rolling-circle amplification, and our proprietary Avidite technology, the AVITI System generates base calling accuracy that exceeds 90% Q30. And it does so at a cost per gigabase on par with factory-level sequencing, without factory-scale batch requirements.
Element introduces a new 2x75 paired-end sequencing kit for RNA sequencing
To bring all these benefits to scientists who use RNA and single-cell RNA sequencing, Element has now released a 2x75 paired-end sequencing kit. With 1 billion reads per flow cell at an operating cost of just ~$1 per million reads, AVITI will let you run more experiments, sequence more cells, or sequence more deeply so you can do more science with your budget.
Brief Overview of Our New 2x75 Sequencing Kits by Isaku Tanida, PhD, Associate Director of Product Marketing
The AVITI 2x75 sequencing kit is a nominal 150-cycle reagent kit that can be used for SE or PE sequencing with an actual cycle capacity of 184 cycles to allow for sequencing both index and bar code sequences. Identical to the larger AVITI 2x150 Sequencing kit, the AVITI 2x75 Sequencing kit offers the same 1B read capacity, making it ideal for counting assays on a cost-per-read basis.
The AVITI System joins the 10X Genomics Compatibility Partner Program
AVITI fits seamlessly within the NGS ecosystem, as shown through its many application partnerships, including 10X Genomics. In fact, Element is only the second-ever member of the 10X Genomics Compatibility Partner Program for Chromium Single Cell 3’ Assays.
Figure 1: Summary of the 10x Chromium Single Cell workflow using the Element AVITI System
As part of the partner program validation, the AVITI system was used
to sequence a control PBMC sample at both small (1,000 cells) and large
(10,000) population scales. Data from the AVITI system met or exceeded
all required CPP performance criteria for both scales, including
standards for barcode validity, mapped reads, reproducibility, and mean
reads per cell. You can learn more details by downloading and reading our application note.
New tools for custom library preparation with the Adept Library Compatibility Kit v1.1.
At Element, increasing access to sequencing goes beyond affordability
– we believe that flexibility is just as important. To provide even greater flexibility
within the AVITI workflow, Element is now enabling users to pursue
custom library sequencing through its launch of a new Adept Library
Compatibility Kit v1.1 and a new Adept Custom Oligonucleotide Buffer Set
kit.
Adept v1.1 features a universal qPCR solution that enables the
quantitation of custom libraries before sequencing, as is currently enabled for standard AVITI
System workflows. The corresponding Adept Custom
Oligonucleotide Buffer Set kit allows users to introduce custom primers
in a separate buffered solution set vs. a spike-in approach. This means
custom primers no longer need to be mixed with primers from the standard
workflow.
Be on the lookout for more exciting new products that further extend
the capabilities of the AVITI System; the best investment in a
sequencing workflow you can make.
Did you know that over 300 million people worldwide live with a rare disease? A "rare disease" is classified as occurring in fewer than 1 in 2,000 people, but collectively, with over 7,000 distinct rare genetic diseases identified, they are common. Rare disease patients and their families can face painful diagnostic odysseys, but trio sequencing can help.
The Unique Challenge of Rare Disease
Families impacted by rare diseases can face tremendous challenges getting help. Symptoms typically manifest in early childhood, are frequently severe, and are often nonspecific or obscure. By definition, their rarity means a typical doctor will not encounter more than one case of a given disease in their entire career. A patient's diagnostic odyssey can last years and include multiple specialists, tests, invasive procedures, and misdiagnoses. The emotional and financial toll on the patient and their family is very high.
In recent years, whole exome sequencing (WES) and whole genome sequencing (WGS) have offered families a shortcut to this diagnostic odyssey by identifying the critical genetic mutation that causes disease.2,3 The success rate of WGS can be improved by sequencing the parents alongside the patient.4 Trio sequencing identifies inheritance patterns, revealing cases where both parents are heterozygous carriers, and the child is homozygous. It is also more effective at uncovering de novo variants arising exclusively in the child, as the parental genomes can be used as healthy controls. Since every individual carries between 40,000-200,000 rare variants present in less than 0.5% of the population but only ~70 de novo variants, having parental genomes can significantly reduce the list of candidate causal genes that must be reviewed. The downside of trio sequencing is that with triple the genomes to sequence, there's triple the cost.
A New Approach to Trio Sequencing
The Element AVITI™ System, our mid-throughput DNA sequencing platform, offers a new way to do trio sequencing while lowering costs. In proof-of-concept work, we aimed to demonstrate that a complete trio study on the AVITI benchtop system can be performed for only $1,680 in sequencing reagents.
Typically, WGS sequencing requires 35x coverage (360 M read pairs) to generate robust variant calling data. That translates into approximately 1 billion read pairs for a trio – exactly matching the recently upgraded AVITI performance specification. However, our partners at Google DeepVariant recently published a manuscript showing that parental coverage can be reduced in trio sequencing while maintaining high variant calling accuracy and excellent de novo variant calling capability.5
To add robustness to the experimental design, we decided to test pooling the trio samples such that the parent library concentrations were half that of the child library. We outlined a complete trio sequencing workflow working with leaders in next-generation sequencing (NGS) for rare disease research. We paired the Roche HyperPlus kit with the Element Elevate™ Library Preparation Workflow for library prep and selected Google DeepTrio for variant calling, following reference alignment via BWA-MEM. The DeepVariant team has developed an Element-optimized version of their neural network-based algorithm. Genoox Franklin was selected for variant interpretation and reporting. Genoox provides an easy-to-use graphical user interface that includes variant annotation and supporting publications, allowing you to easily connect with other researchers in your community with the same disease focus. DeepTrio and Genoox are widely used in leading institutions, providing low or no-cost options for their analysis pipelines. The complete sequencing and analysis workflow is shown in Figure 1.
Figure 1: End-to-end workflow for rare and undiagnos3ed disease (RUGD) trios
Partnering with UC San Diego to Find the Drivers of Rare Eye Disease
We partnered with rare eye disease experts UC San Diego Professor
Radha Ayyagari, PhD, and her graduate student, Pooja Biswas, to
demonstrate the potential of this application to reduce costs without
sacrificing efficacy.
You may recall that we previously worked with Professor Ayyagari's
work was presented at our launch event, where Element sequenced DNA on
the AVITI system from 10 subjects known to suffer from retinitis
pigmentosa. We leveraged partnerships with Roche, Sentieon, Genoox, and
Fabric to identify high-confidence candidates in five cases—three of
which were previously unsolved. Pending clinical validation, this
represents a 50% solve rate. Watch the video to learn more about the specific variants that were discovered.
For this new collaboration, we took up yet another rare eye disease
case where none of the family had previously been sequenced, aiming to
explore what WGS of the full mother, father, and child trio might
reveal, using our newly outlined workflow.
New Hope for an Answer
The run generated over 1.1 billion read pairs and 326 GB of
sequencing data, yielding greater than 20x in each parent and 50x in the
proband, with 94.6% of the data at Q30 of higher.
Google DeepTrio variant calling and Genoox Franklin Workbench
interpretation identified a high-priority homozygous stop gain in the
CERKL gene. In this case, both parents are heterozygous carriers. The
variant is classified as pathogenic ClinVar and is associated with
retinitis pigmentosa, a rare eye disease consistent with the patient
phenotype. Pending confirmation via clinical sequencing, the variant is
likely causal.
Figure 2: Genoox Franklin reports a CERKL gene mutation as a highly prioritized variant.
Figure 3: IGV display showing both parents are heterozygous carriers while the proband is homozygous.
A Promising New Path to Democratize Trio Sequencing
This proof-of-concept study shows the AVITI system's promise for
democratizing trio sequencing access via an affordable benchtop system
and cloud-based analysis solutions. Much work remains to be done, and we
are already pursuing more extensive studies across a broader range of
disease contexts where de novo variants are more frequently responsible.
To hear more details about this study, watch Semyon Kruglyak, PhD, Vice President of Informatics at Element, present the team's results at AGBT below.
]]>urn:uuid:ce20d78e-4dc3-43cd-97ad-b4ed020fa671What Is Element LoopSeq™ Long-Read Sequencing Chemistry?2022-08-23T08:00:00-07:002023-05-19T09:02:48-07:00Tuval Ben-Yehezkel, PhDtuval.benyehezkel@elembio.comA High Level Overview of Element LoopSeq Long-Read Sequencing Chemistry
Synthetic solutions to long-read problems
Over the past two decades, advances in next-generation sequencing (NGS) technology have significantly increased throughput to offer more genomic data at a lower cost. This increase, however, comes at a cost of shorter read lengths and, by extension, long-read applications. Traditional short-read technology reads molecules base-by-base and the resulting cumulative deterioration of signal over hundreds of cycles restricts results to a few hundred nucleotides. To overcome this limitation, molecular biologists devising early synthetic long-read (SLR) library prep methods used molecular tags and limiting dilution when preparing short-read libraries. The preserved tags were then used to informatically piece together longer sequences.
Several years later 10X Genomics first commercialized SLR, introducing a microfluidic device that increased throughput and streamlined the workflow. LoopSeq long-read library prep pushes the technology even further, obviating the need for microfluidics and expanding applications.
Unlocking a range of long-read applications
In contrast to SLR predecessors—whose per-molecule coverage was too low for true single-molecule long-read sequencing—LoopSeq targets complete coverage of long molecules. This novel technology assembles continuous long reads from single molecules, generating data identical in type to the data that legacy long-read technology companies such as PacBio and Oxford Nanopore generate.
The ability to assemble continuous long reads dramatically broadens the scope of NGS applications to embrace transcriptome, microbiome, and immune repertoire sequencing, to name a few. Moreover, LoopSeq chemistry eliminates the requirement to physically compartmentalize DNA molecules with limited dilution, enabling a more flexible workflow that does not require dedicated instrumentation. Figure 1 details the end-to-end workflow.
Figure 1: The main steps of LoopSeq long-read sequencing from library prep through analysis
Lower error rates, higher accuracy
Arguably the biggest advantage LoopSeq offers compared to legacy SLR
technology is higher accuracy and lower error rates. LoopSeq uses raw
data (short reads) that typically have better error rates than the
legacy long-read counterparts. LoopSeq then applies consensus-based
error correction at each position of a long read based on many
independent short reads that cover each position in a long read. The
resulting long reads are extremely accurate, as demonstrated in Figure
2.¹
A key observation in Figure 1 and the study that produced it is the
nonlinear relationship between the length of a long read and the
probability that it is error-free. A long molecule is exponentially, not
linearly, more likely to contain an error compared to a shorter
molecule. As a result, the low error rate for 1.5 kb LoopSeq reads leads
to roughly 50% more error-free reads compared to PacBio. For 5 kb
reads, the rate of error-free reads increases to 500% more error-free
reads. A roughly three-fold increase in length leads to a 10-fold
increase in error-free reads, making LoopSeq accuracy increasingly
important with read length and improving confidence in results.
Figure 2: A comparison of error profiles from sequencing the same samples using LoopSeq, PacBio, and Oxford Nanopore technologies²
More specifically, the study examined how to sequence complete 1.5 kb
bacterial 16S DNA, 2.3 kb fungal 18S-ITS, and entire 5 kb bacterial
ribosomal DNA (rDNA) clusters with a single long read to achieve
extremely high accuracy. The study also addresses how accurate long
reads enable species- and strain-level microbiome classifications.
Another study of LoopSeq error rates for the transcriptome space
provides a comparative analysis of human messenger RNA (mRNA) spanning
multiple sequencing technologies.² In this study, the authors find that
LoopSeq error rates are significantly lower than other short- and
long-read technologies across all error types, as shown in Table 1.
Crucially, cancer progression involves isoform switching, in which
specific clonotypes evolve to express isoform-specific single-point
mutations. This tiny variation in the sequence of isoforms leads to
vastly different phenotypes, a discovery made possible only by the low
error rates inherent in Element LoopSeq long-read technology.
Error Type
PacBio-CCS RNA
ONT-2D RNA
Illumina RNA
LoopSeq RNA
LoopSeq DNA
Match
9.83E-01
8.66E-01
9.95E-01
9.99E-01
1.00E+00
Substitution
1.30E-02
5.50E-02
4.15E-03
3.48E-04
1.48E-04
Insertion
8.70E-04
3.12E-02
3.84E-04
2.06E-04
2.15E-06
Deletion
3.40E-03
4.79E-02
4.80E-04
2.69E-04
2.39E-06
Sum error
1.72E-02
1.34E-01
5.02E-03
8.23E-04
1.52E-04
Table 1: Comparing error rates across diverse sequencing platforms²
In sum, the ability to obtain highly accurate long reads has been an
enduring challenge. The enhanced resolution of LoopSeq long reads
enables interrogation of long DNA and RNA molecules using previously
inaccessible methods to continue the growth and variety of applications
across the field of genomics.
To learn more about Element LoopSeq and the benefits of our exclusive offering of both short- and long-read sequencing, visit Element LoopSeq Long-Read Workflows.
¹ Callahan, Benjamin J., Dmitry Grinevich, Siddhartha Thakur, et al.,
“Ultra-accurate microbial amplicon sequencing with synthetic long
reads,” Microbiome 9, no. 130 (June 2021): https://doi.org/10.1186/s40168-021-01072-3.
² Liu, Silvia, Indira Wu, Yan-Ping Yu, et al., “Targeted
transcriptome analysis using synthetic long read sequencing uncovers
isoform reprograming in the progression of colon cancer,” Communications Biology 4, no. 506 (April 2021): https://doi.org/10.1038/s42003-021-02024-1.
]]>urn:uuid:ff765481-c7f7-4f33-9ffd-1f168a735db8Measuring the Accuracy of Element AVITI™ Sequencing Data2022-07-13T10:00:00-07:002025-09-26T08:48:28-07:00Semyon Kruglyak, PhDsemyon.kruglyak@elembio.com
At Element Biosciences, we are committed to setting new standards for accuracy while driving applications that leverage that accuracy to achieve new or more efficient results. The Element AVITI System combines exceptional accuracy with a high number of reads and low operating costs.
In this article, we describe the accuracy of the AVITI System and how that accuracy is measured.
What Are Q-Scores?
The field of genomics has developed and evolved tremendously over the last 25 years, but a few central pillars have remained largely intact.
Quality scores (Q-scores) based on the Phred scale are a great example. In 1992, the concept of Q-scores was proposed as part of the SCF file format. A formal proposal of a numerical scoring method soon followed in a 1995 Nucleic Acids Research paper authored by James Bonfield and Rodger Staden at the University of Cambridge MRC Laboratory. The new ability to generate machine-readable data from sequencing traces for the ABI 373A and Pharmacia ALF instruments motivated the paper.
Until the arrival of automated sequencing, base calls were subjective and based on autoradiographs. Bonfield and Staden proposed the novel idea that base-calling algorithms can produce numerical estimates of accuracy at each base, building on earlier error correction methods. Their simple concept flourished, becoming the cornerstone of competitive positioning for a variety of sequencing platforms generating billions of reads per run.
How Are Q-Scores Determined?
Modern FASTQ files encode Q-scores for each base in each sequencing read.
The field has generally settled on Q30—or one error in every 1000 bases—as a reasonable accuracy standard for sequencing. Some long-read platforms consider one error in every 100 bases (Q20) as a reasonable standard. Although Q20 and Q30 are often mentioned, base call values of Q40 and above have not been broadly considered due to the accuracy limitations of current sequencing technologies.
Accuracy is typically described in terms of Q-scores, which are the log-transformed error probabilities.
Table 1 presents the interpretation of common Q-scores. The quality specification for the AVITI System is at least 90% Q30 computed on a 2 x 150 bp run. The minimum output on each of two independently operated flow cells is 800 million read pairs. In practice, both the accuracy and number or reads specifications are often significantly exceeded. We begin by defining Q-scores and explaining how they are assigned. We then demonstrate that the Q-scores accurately represent the underlying data quality.
Table 1: Interpretation of Q-scores
Quality Score
Interpretation
Q10
1 error in 10 bases
Q20
1 error in 100 bases
Q30
1 error in 1000 bases
Q40
1 error in 10,000 bases
Q50
1 error in 100,000 bases
How Element Biosciences Assigns Q-Scores
To assign Q-scores, Element follows the methods of Ewing et al. but with custom predictors. Briefly, we generated 20 runs of whole-human sequencing (WGS) data for training and leveraged Covaris sheared, PCR-free library prep to limit upstream errors. The base calls were labeled as either correct or as erroneous based on alignment data. The base calls and labels served as input for the training process. In turn, the output of the training process was a table that maps predictor values to Q-scores. The table was applied to an independent run (not used in the training) to generate Figure 1 (seen below), which illustrates the run. Download the run from our Sequencing Datasets page here.
Figure 1: Predicted and observed quality scores for a 2x150 bp human genome sequencing run
The histogram shows that most of the base calls exceed Q40, with Q44 being the most frequent assignment.
The blue dots show that the predicted Q-scores match the recalibrated Q-scores well.
To obtain the recalibrated (empirical) Q-scores, GATK Recalibrator is paired with publicly available known-sites files to prevent bases overlapping variant positions from being counted as sequencing errors. Figure 2 details how GATK Recalibrator was applied to generate the data.
Figure 2: Application of GATK BaseRecalibrator to the data
Data Filtering
In addition to showing the Q-scores and verifying accuracy, describing any data filtering is also important:
First, we eliminate read pairs that have poor quality in the early cycles of either read. This procedure typically removes ~5% of the overall data. For the cited sequencing run, the number of read pairs that pass filter is ~1.02 billion, far exceeding the 800 million read pair specification.
Second, we trim adapter sequences by matching the ends of the reads to the known adapter sequence. The amount of filtering this step performs depends on the insert length distribution and read length. Both the adapter-trimmed and untrimmed data are available. Notably, we do not perform any filtering based on alignment or any other knowledge that secondary analysis generates.
Achieving Q40+ on AVITI
In conclusion, Element data generated from PCR-free libraries has a high proportion of Q40 data. The assigned Q-scores are accurate across the entire range of scores as determined by open source third-party tools. Minimal filtering is applied to the raw the data, and our website offers the trimmed and untrimmed data in FASTQ format.
To learn more about what the Element AVITI System can do for you, reach out to our team today.
]]>urn:uuid:db0cb4ad-61ff-48bc-8a81-d443856870f9Advances in Long-Read Sequencing with Short-Read Next-Generation Sequencers2022-07-06T16:39:00-07:002023-05-19T09:09:35-07:00Tuval Ben-Yehezkel, PhDtuval.benyehezkel@elembio.com
The Ascent of Short-Read Next-Generation Sequencing
During the past two decades, short-read next-generation sequencing (srNGS) has expanded the science of genetics far beyond what became possible using Fred Sanger’s first-ever sequencing technology (AKA Sanger sequencing), developed in 1977. The impact of srNGS has been so far-reaching that “genetics” was renamed to “genomics” to account for the vast increase in genetic information the new technology generated compared to its predecessor.
Since its early deployment twenty years ago, srNGS has been on a steady trajectory of generating more data for fewer resources. Genomes that took years to sequence can now be sequenced in hours, revolutionizing genomics research and development, with widespread impact on everyday life.
srNGS now permeates every domain of the life sciences and healthcare, enabling advancements in vaccine and drug development, better monitoring and early detection of disease, as well as numerous other contributions on a broad application spectrum ranging from increasing crop productivity to pushing the boundaries of pre-natal diagnostics, to name a few.
Challenges and Limitations of Short-Read NGS
Two decades of srNGS have also revealed many genomics challenges that short reads cannot fully resolve. Domains such as genome, transcriptome, microbiome, and immune repertoire sequencing are a few areas where short-read lengths have limitations. The wide availability of inexpensive and accurate short-read sequencing has provided a foundation to appreciate that the true complexities of the genome and transcriptome will benefit from higher and higher resolution. Improvements in resolution are most recognized in higher accuracy sequencing, longer read lengths, or ideally, both.
Many biological questions cannot be fully resolved with short reads, regardless of how many of them we throw at the problem. Genomics will increasingly require alternative technologies that push the boundaries of existing srNGS. Making this trend even more severe are two decades of short-read thinking (see the Law of the Instrument), conditioning scientists to view problems through the lens of the short-read sequencer. This trend results in biases in how scientists think about experiments in genomics, primarily seeking to address questions that can be answered with short reads and neglecting questions that short reads cannot answer.
In other words, we are devising scientific questions that fit the existing sequencing technology instead of devising sequencing technologies to fit the scientific questions we want to ask.
Advancements in Long-Read Technologies
Fortunately, genomic technologies are diversifying in technology platforms that allow native long-read sequencing and applications that leverage short-read output to develop long-read resolution through creative molecular biology and bioinformatics.
Native long-read platforms offer unparalleled opportunities to sequence tens of thousands to millions of bases in a single read. Tradeoffs for the longer reads can be lower per-base accuracy or higher cost per read compared to short-read technologies. Application-based methods that leverage short read platforms’ accuracy and cost advantages are also advancing.
Multiple new sequencing technologies are emerging in the long-read and synthetic long sequencing space, enabling scientists to uncover previously inaccessible genetic information. The opportunities these technologies present are everywhere and are positioned to accelerate advancements in antibody development through the sequencing of full-length antibodies, drug target identification through the sequencing of disease-specific transcription isoforms, and the development of more effective viral vaccines through the characterization of viral quasi-species, to name a few examples.
Learn More About Long-Read Sequencing with Loop Genomics
To learn how you can get a head start in your research by integrating advanced sequencing technologies like synthetic long-read sequencing, visit us at the Loop Genomics’ website's Learn section to read peer-reviewed publications, listen to webinars, and read tech notes about Loopseq™, our emerging synthetic long-read sequencing technology.
Join us and the ever-growing community of scientists embracing new long-read sequencing technologies and participate in bringing about the future!
]]>urn:uuid:16bcea83-a184-41ce-a527-a7d16323e2a6An NGS Replacement for Sanger Sequencing: A Protein Engineering Case Study - Video and Q&A Transcript from AGBT 20222022-07-06T12:30:00-07:002023-05-19T09:34:07-07:00Matt Kellinger, PhDmkellinger@elembio.comAGBT 2022 Spotlight Talk - Presented by Matt Kellinger, PhD
Q: How is the long-read Sanger sequencing achieved? If you have a barcode ligated there, once you fragment, do some pieces not have the barcode?
A: In traditional approaches, that is what happens. Some of the cleverness of this method comes through in the molecular biology and it is worth jumping back into this distribution reaction. If the gene of interest is 2KB or 5KB, what happens in that step is the barcode on that end is inserted uniformly all along that gene. You have 1000s of copies and they each have a unique address and then you have a known barcode next to some part of the gene that you can access. That is how you do the short read to long read jump. You actually sequencing maybe only 150 bases, but you are doing it at all these different places along the gene and you have the barcode for reassembly.
Q: Even if you distribute uniformly, some of your short reads won’t have the UMI?
A: Actually they will, the way the fragmentation is done. It turns out that the UMI winds up at the beginning of every read; then you can aggregate by UMI and then throw it in your assembler.
Q: Have you tried to use a short-read instrument to do long reads and to see performance compared to performance with PacBio or Nanopore?
A: This system is short-read technology. Today it gives you 2x150 for a total read length. You can play with insert size to get it longer but you will still end up with 2x150 base pair reads. So for this application, that would mean a lot of missed content in the middle, if everything is not individually barcoded, reassembly would not happen. So that is what this assay overcomes with some molecular biology - a way to get to a synthetic long-read limited by long-range PCR – 20Kb is typically the end of that size. In terms of comparison to PacBio or ONT, there are several publications that address this topic.
Q: One of the constant challenges with Illumina from the very beginning is the absolute requirement for high complexity. Is that a similar requirement for Element’s AVITI? Or now, can we now simply take homopolymer stretches, a whole bunch of clones with exactly the same sequence and put them on with one change within a thousand clones and directly sequence it? Or do we need something like this that is going to randomly fragment to give us that complexity?
A: We have low diversity methods which are implemented, what we require is a reasonable diversity within the first 5 cycles. We are working on eliminating that requirement in our own Elevate prep, but if you are using compatibility, we do require that diversity within the first 5 cycles, after that it is pretty robust.
Q: How low can diversity be outside the initial cycles?
A: So we should be good with arbitrarily low. We haven’t worked it out extensively, but we would be excited to try your use-case and then figure out if we need maybe a 5% PHiX spike-in or if we are good to go.
Q: What is level of sample multiplexing can be achieved?
A: It is a single barcode on the front but there are 3 layers of barcoding that can happen so the multiplex levels can get high. You can have a well-based barcode which is this one, but then you can have plate-based barcodes so you don’t need endless well barcodes and then you can have barcode for the NGS run itself. So in a 96 plex sequencing run, one index could be dedicated to an application like this and inside that one index, you have 2 more layers of indexing which can let you put 1000s of samples inside that one NGS run.
]]>urn:uuid:c2fd8a32-fce1-49d3-9c47-07f82b56e508Uncovering the Meta-Transcriptome with Long-Read Sequencing - Video and Q&A Transcript from AGBT 20222022-07-06T12:00:00-07:002023-05-19T09:39:08-07:00Shawn Levy, PhDshawn.levy@elembio.comAGBT 2022 Spotlight Talk - Presented by Shawn Levy, PhD
Q: Theoretically, once you make the library, whether you sequence with Illumina or AVITI, both are short-read technology and both should come out with similar quality, but you are pointing out that AVITI is better, why?
A: Importantly, the short-read sequencing which was done on Illumina was done short-read only. The Loop Seq was not applied to that short-read – a key differentiation. But to more appropriately answer your question, with the Loop Seq library, if we had sequenced that on AVITI and Illumina, the major conclusions would be the same. The differences would lie in some of the short-read performance differences – e.g. cost/Gb, relative base quality performance. It is really 2 different considerations.
Q: Have you tried the Loop technology on 10X Genomics cDNAs? Sometimes people use Nanopore to get full-length DNA of the single-cell libraries. Can you imagine whether we would be able to do this with Loop?
A: Conceivably it should be workable in a similar fashion, but it isn’t something we have experimentally looked at.
Q: Do you read the barcodes separately like Illumina or is it included or is it included in the 150 reads?
A: The UMI sequence is the initial part of R1. But then there are other indexes that can be used for sample and for plate. So you have multiple tiers of indexing capability. So the answer is both. It is captured in both cases.
]]>urn:uuid:7b3e21da-9cf8-49b8-bd49-0f8c7cf9c2ceCost-Effective Trio Sequencing - Video and Q&A Transcript from AGBT 20222022-06-29T16:30:00-07:002023-05-19T09:42:57-07:00Semyon Kruglyak, PhDsemyon.kruglyak@elembio.comCost-Effective Trio Sequencing - Presented by Semyon Kruglyak, PhD, at the AGBT General Meeting on June 7, 2022
Q: In regards to library prep, is this closer to HyperPlus?
A: We work with HyperPlus and HyperPrep, but we are
fairly agnostic. There’s a long list of supported third party providers
that we are compatible with. View full list here.
Q: Is there a way to compare the same genomes that you can run on, for
example, Illumina? It seems like there are similar workflows.
A: Yes, we use our Elevate™ library prep workflow with Roche KAPA. But if you want to do a transposon-based prep, that will be OK too.
Q: In regards to the rate of switching from one spot to another between Read-1 and Read-2 — how have you dealt with getting the optics realigned exactly in the right place?
A: We generate our feature set based on Read-1 and then the features don’t really move. You can correct for the various optics and you can map. Because this is a random flow cell, there’s only one unique mapping — so you take the images of Read-2 and very accurately map them back onto the Read-1 map that was previously generated. From what we have seen, switching spots between Read-1 and Read-2 is extremely rare.
Q: Looking to the future, where is Element Biosciences heading?
A: It’s relatively new technology, so there’s a lot of headroom in all dimensions. We promise 800 million read pairs but the run shown here is 1.1 billion. We are working to determine which improvement directions would be of greatest value to the customer.
]]>urn:uuid:c70b38a4-5041-4680-80ab-bbc74e44c592Q40+ on AVITI - Video and Q&A Transcript from AGBT 20222022-06-29T16:00:00-07:002023-05-19T09:47:15-07:00Semyon Kruglyak, PhDsemyon.kruglyak@elembio.comQ40+ on AVITI - Presented by Semyon Kruglyak, PhD, at the AGBT General Meeting on June 7, 2022
Q: How hard is it to hit the density target when deciding on library concentration?
A: We have a quantification step that helps. You may see some variability when first trying, but then you dial it in. We also accept a fairly high range for the density.
Q: Is it a patterned flow cell?
A: It is a random flow cell.
Q: What are the read lengths?
A: Our data is generated from 2x150 for this talk. You can of course go shorter.
Q: What types of libraries have you run?
A: We have a long list of library types through a broad ecosystem of partners — whole human genome, RNA, single cell, exomes, panels, and more.
Q: What do you see as your position in the market?
A: We want to lead with a quality, while offering a cost-effective benchtop solution. You can do runs in your own lab with full flexibility and you don’t have to send out to get the economies of scale.
Q: What is the price of the instrument?
A: The sequencer is $289,000 and a 2x150 kit is $1,680 (sequencing reagents and flowcell).
Q: Is the current data filtered?
A: There is minimal filtering. If a feature has low quality in early cycles, we filter it. In general, we filter 5% of our data or less, and the rest goes into the FASTQ file that you download. There’s no other manipulation beyond those early cycles of R1 and R2, with the exception of optional adapter trimming.
Q: Are the errors random?
A: The errors seem mostly random. Since we are fairly early in our review it’s something that we’re still looking into.
Q: How do you handle low diversity?
A: We require high diversity in the first few cycles of Read-1 so we can find where all the features are. Beyond that, we have a quite robust low diversity pipeline that handles the type of challenges that low diversity presents.
Q: Have you sequenced bisulfite converted DNA (i.e. methylseq)? And does data from methylseq have great quality scores as well?
A: Yes, we have, and they seem to work pretty well, though density is somewhat reduced. We would love to partner with someone to optimize performance on these libraries.
Q: How long does it take to run the 2x150?
A: Just under 48 hours.
Q: Do you support dual indices?
A: Yes, we support dual indices and support two types of library prep. If you have your own linear library, we will take it and apply our compatibility kit and then run it with however many indices you have. If you use our Elevate library, then we have unique dual indices for 96 plex and many more if you do not need UDI.
Q: Are dual indices needed on this platform?
A: Although our amplification methodology does not appear to lead to index hopping, they may be considered for other reasons. For example, if your index plate is all synthesized together, you can have oligo contamination — leading to index misassignment. This is something that we’re currently exploring.
Q: What’s the duplicate rate?
A: We are seeing very low duplicate rates. Typically sub 1%.