Single-cell RNA sequencing (scRNA-seq) is a transformative technology in biology that unlocks the complexity of cellular heterogeneity, developmental dynamics, and disease mechanisms with unprecedented resolution. Continued advancements in single-cell tools that better characterize gene expression from individual cells have further improved our ability to decipher these complexities, though technical limitations within the field of next-generation sequencing have restricted our ability to further explore this space.
Impacts of Homopolymer Regions on Sequencing Single-Cell Libraries
Challenges with standard SBS sequencing technologies have limited the use of paired-end sequencing of standard 3’ scRNA-seq libraries because of challenges associated with sequencing through homopolymers. Using 3’ priming methods for RNA-seq library generation, such as those provided by some 10x Genomics protocols, result in a library molecule where a portion of the poly-A tail is retained between the R1 sequencing primer and the coding region of the RNA. During standard clonal amplification on SBS sequencing platforms, these molecules undergo PCR using enzymes that introduce considerable error in these regions and result in clusters of molecules with a diverse length distribution of adenosine stretches. Sequencing through these stretches results in considerable phasing that can be seen in subsequent basecall results, yielding data of questionable utility. Because of this, standard sequencing scRNA-seq sequencing workflows rely solely on transcript data generated from the 5’ end of the library insert.
Exploring the Advantages of Avidite Base Chemistry (ABC) Sequencing in scRNA-seq
Element Biosciences has leveraged a different approach to amplifying library molecules on the surface of the flow cell, using rolling circle amplification (RCA) to generate polonies. As shown in the image below, a single molecule is used as a template by enabling the polymerase to extend the surface primer continuously through an isothermal process. By using a single template to create thousands of copies, error proliferation of difficult regions seen from PCR-based methods doesn’t occur, resulting in a highly uniform population of homopolymer length.
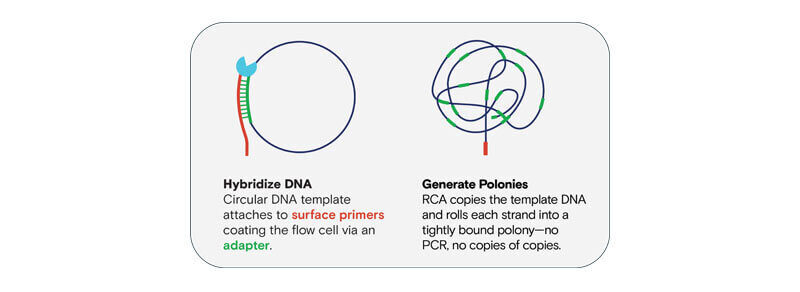
Download our ABC Sequencing infographic to learn more.
A team of researchers from the University of Utah School of Medicine noted the unique accuracy advantages enabled by ABC sequencing technology and evaluated the potential impact of paired-end sequencing to standard 3’ scRNAseq libraries in single-cell transcriptomics. The use of an AVITI™ system to overcome limitations of homopolymer regions was evaluated in a recent preprint posted to BioRxiv titled Improved characterization of single-cell RNA-seq libraries with paired-end avidite sequencing. The authors hypothesized that, by obtaining information from both sides of the RNA sequence, errors from misaligned reads and those that were unable to be uniquely mapped using single end data would be diminished during analysis. Additionally, by reading from the 3’ end of the transcript, polyadenylation site assignment could be more accurately defined and quantitated.
Increases in accuracy relative to SBS sequencing chemistry were well documented in the 2023 Nature Biotechnology publication Sequencing by avidite enables high accuracy with low reagent consumption, though this article highlights the first application of ABC towards true paired-end scRNAseq.
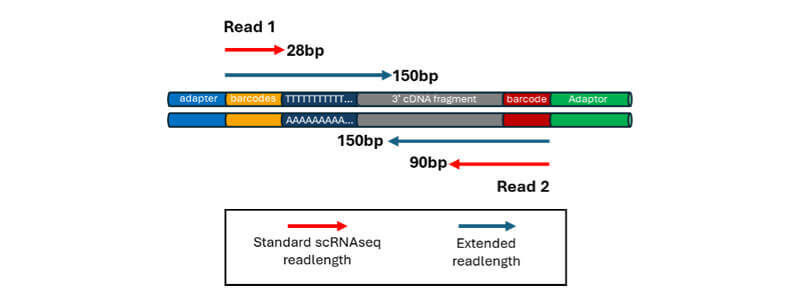
Paired-End Sequencing of scRNA-seq Libraries
The authors generated 5 scRNAseq libraries using the 10x Genomics Chromium 3’ workflow and sequenced those libraries on both a NovaSeq6000 and an Element AVITI. Unlike standard 10x library sequencing schemes where 28bp are generated from read 1, allowing barcode and UMI identification only, the full 150bp read was generated from both platforms. As a secondary point of study, read 2, normally sequenced to 90bp, was also extended to 150bp.
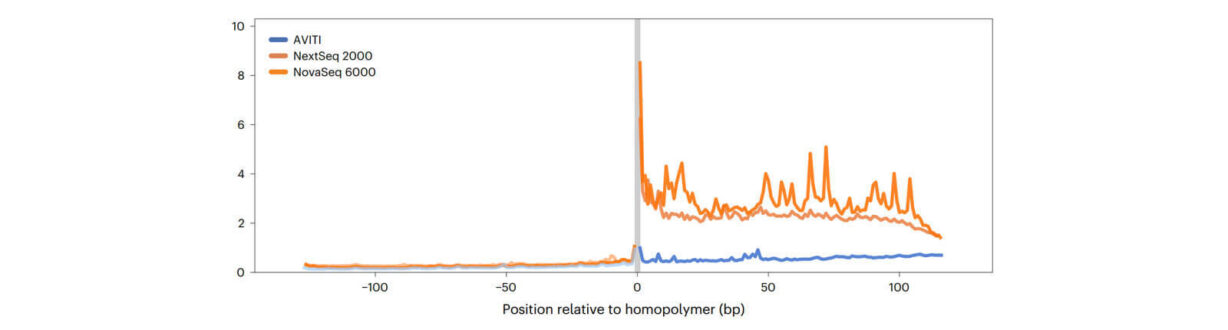
Not surprisingly, results from read 1 generated on the NovaSeq experienced a significant drop in quality following the poly-A region where no measurable decrease in quality was observed with AVITI data. This further translated to an approximate 4x increase in unique alignments to the reference relative to NovaSeq.
Further analysis of paired end reads compared to standard single-end scRNAseq data, results indicated that, while the use of a second read did increase unique mapping statistics, the overall alignment rate was similar. The authors indicated the likely cause of the reduced impact of paired reads was short library length, though data did show a significant increase in base-pair precision of polyadenylation site assingement.
The Impact of Sequencing through Polyadenylation Sites
Several methods exist to predict polyadenylation sites based on RNAseq data, many are described in a 2023 publication, A Survey on Methods for Predicting Polyadenylation Sites from DNA Sequences, Bulk RNA-seq, and Single-cell RNA-seq. Advances in modeling, such as the use of deep learning algorithms, have improved the ability to do so, though all of these tools are based on the assumption that emperical sequence information from 3′ ends of mRNAs for direct profiling of poly-A sites remains a technical challenge. Although methods such as TAIL-Seq and PAT-Seq have been developed to do so, the workflows are considered to be extremely complicated and costly. To the contrary, paired-end sequencing of scRNAseq libraries using the AVITI enables a scalable, simple alternative to providing true emperical characterization of polyadenylation in parallel with single-cell gene expression analysis.
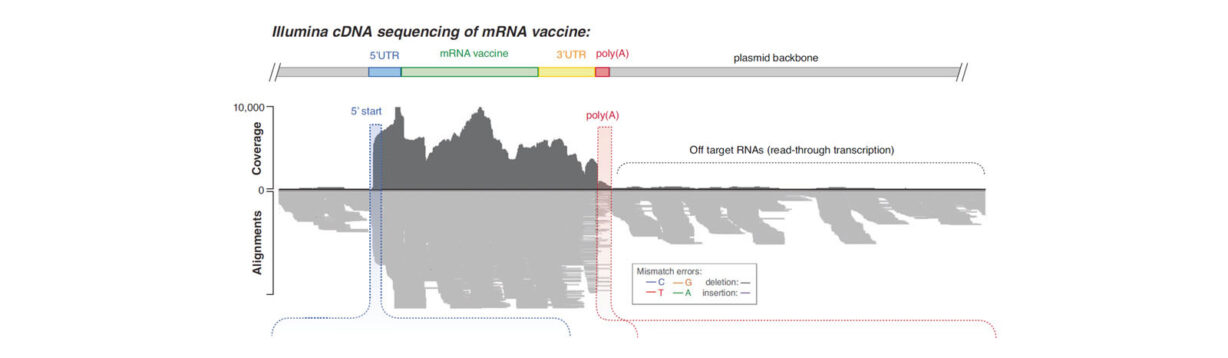
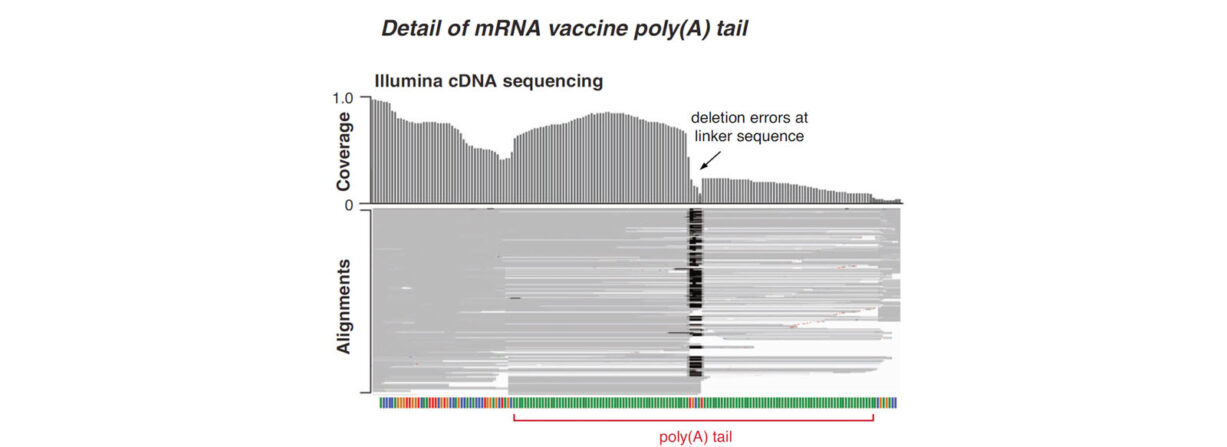
The ability to sequence through poly-A tails with high accuracy has implications beyond those noted by the authors. An area that may benefit from these advancements include the evolving field of mRNA vaccines, where accurate methods to QC plasmid templates containing the transcript sequence are required during the manufacturing process. Recent publications propose methods using NGS to do so, though a 2023 publication in Nature Communcations presented data highlightlighting challenges with SBS chemistry in accurately sequencing through the poly(A) region, a critical measurement required in mRNA vaccine QC.
Single-cell gene expression analysis has proven to be a breakthrough technique, ushering in a new era of discovery through profound insights into cellular heterogeneity, developmental processes, disease mechanisms, and therapeutic interventions. The ability to measure additional regulatory mechanisms of transcription like alternative polyadenylation has the potential of providing further understanding of cell-to-cell variability at the RNA level, though emerging single-cell multiomic approaches can further expand our view of systems biology beyond gene expression. Cytoprofiling capabilities of the Element Biosciences AVITI24™ platform have the ability of detecting and localizing RNA and proteins within cells and combine intra and extracellular morphology phenotypes to enable a more comprehensive view of individual cells at an unprecedented level. With the introduction of advanced tools like AVITI24, researchers can now map complex cellular pathways at a resolution that has not been previously obtainable.