The human genome spans about 3 billion base pairs packaged in 23 pairs of chromosomes.1 Approximately 23,500 genes are scattered throughout the genome and bookended by other regions of DNA. Collectively, these genes include about 180,000 protein-coding regions called exons, which comprise the exome.1 Encoded within this human complexity are answers to questions researchers have about myriad health conditions, including inherited disease and cancer. With whole-exome sequencing (WES), they do not have to sequence an entire genome to trace phenotypes and identify disease-related variants.
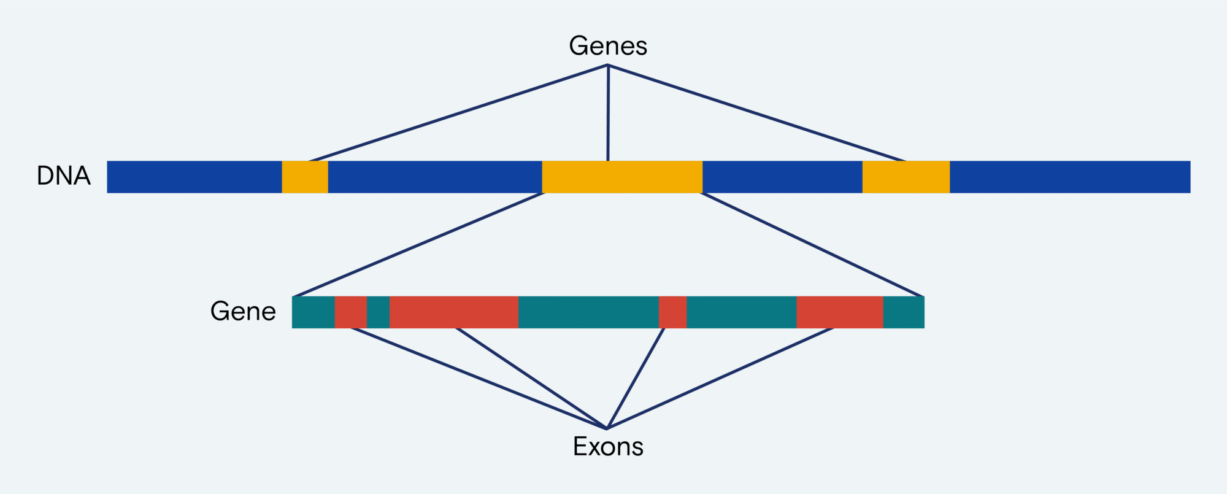
Comprising only 1% of the genome, exons nonetheless harbor 85% of the genetic mutations known to cause disease.2 By isolating and assessing only exons, researchers extract meaningful data from less DNA to save time and money. WES assesses exons for known and candidate variants, asserting utility for population genetics and research of genetic disease and cancer. The abundance of each variant type is wide-ranging—from tens to millions per genome—so sensitive and accurate sequence detection requires optimization across multiple parameters.
Variant | Approximate Quantity |
DNA sequence variants (DSVs) | 4 million |
Single nucleotide polymorphisms (SNPs) | 3.5 million |
Nonsynonymous coding SNPs (nsSNPs) | 13,500 |
Loss-of-function (LoF) heterozygous variants | 100–120 |
Variants associated with inherited diseases | 50–100 |
De novo variants | 30 |
Table 1: Abundance of variants in the human genome1
Targeting the one percent
A WES experiment progresses through the same workflow as most next-generation sequencing (NGS) applications and adds target enrichment:
- Library prep—Prepare a library from DNA input to access a genome.
- Target enrichment—Select the 1% subset of DNA that encodes proteins.
- Sequencing—Read and convert the library into files that contain the genome sequence.
- Analysis—Gain insight from the sequencing data.
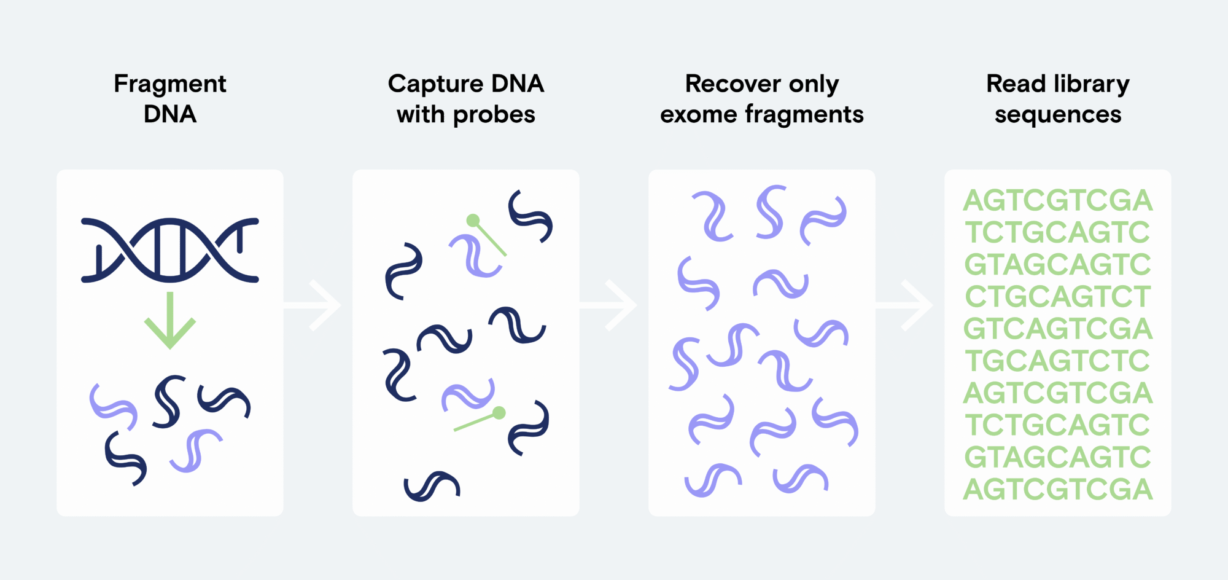
WES is essentially targeted sequencing. Oligonucleotide probes complementary to all exon regions capture relevant DNA for sequencing. Unwanted DNA is washed away. These whole-exome probe panels are readily available through a variety of suppliers. Panels vary in size, hands-on time, automation capability, and compatibility with analysis software and variant databases. When selecting a solution, weighing panel size and completeness against the cost of sequencing required for a panel-appropriate depth of coverage is important.
Deep insights with less commitment
Arrays and whole-genome sequencing (WGS) have similar goals as exome sequencing but differ in breadth of variation detection within a genome, making WES something of a happy medium:
- At one end of the spectrum, arrays detect only variants that are common to many individuals in a population. They also require more input and specialized hardware.
- At the other end, WGS reports all nucleotides in the genome, picking up both coding and noncoding regions and mitochondrial DNA (mtDNA).
For certain applications, the high volume of sequencing data that WGS provides is worthwhile. WGS is, for example, a better fit for discovering novel genes and mutations. However, when detecting coding variants is sufficient, WES provides a faster, more cost-efficient approach. As disease-related variants are highly likely to dwell in protein-coding sequences, targeting the exome costs less than WGS while still yielding many useful variants. Moreover, the smaller dataset that WES generates enables faster, more manageable analysis.
Sorting true variants from false
Like any detection method, WES is vulnerable to false positives and false negatives, making sequencing quality extremely important. Incorrect base calls lead to false-positive and false-negative mutations, a problem that higher sequencing quality with fewer errors to begin with mitigates—as does higher coverage to mask low-probability errors. Therefore, higher quality translates to higher accuracy, in turn distinguishing true variants and enabling reduced coverage that can save on costs or increase throughput. Conversely, researchers have the flexibility to sequence to a greater depth.
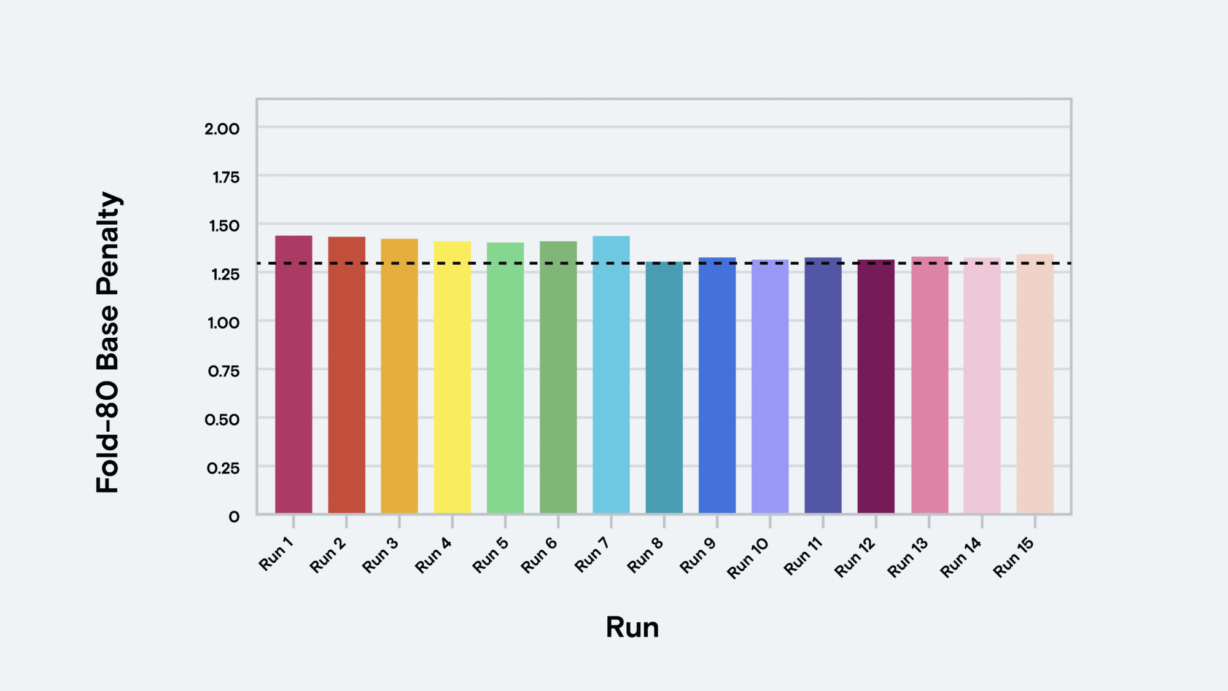
Uniformity of coverage, another accuracy driver, indicates whether target enrichment captured each sequence at roughly equal rates. Minimally captured target sequences compel additional sequencing runs to ensure adequate representation, a consequence represented as fold-80 base penalty: the amount of extra sequencing reads needed for 80% of the targets to reach mean coverage. A low fold-80 base penalty is desirable because it indicates uniform target representation and cost-effective sequencing. A high fold-80 base penalty wastes reads on well-captured targets and requires more sequencing.
Optimal quality and cost
Avidite base chemistry (ABC) further optimizes the WES workflow by offering a high-quality, low-cost alternative to sequencing-by-synthesis (SBS). ABC, which powers the Element AVITI™ System, delivers superior accuracy across a range of coverages.3,4
A study assessing AVITI performance with Twist Exome 2.0 generated 1.1 billion reads at a sequencing cost of only $45 per exome. AVITI exceeded 90% Q30 and achieved nearly 90% Q40 bases with uniform coverage, efficient depth of coverage, and a low duplication rate. Fold-80 base penalty reached only 1.3, demonstrating efficient sequencing that delivered uniform coverage.
Metric | Value |
Assignment rate | 97.4% |
Coverage > 50x | 86.9% |
Duplication rate | 2.7% |
Fold-80 base penalty | 1.3 |
Q30 | 97% |
Q40 | 86.9% |
Read count | 1.1 billion |
Yield (Gbp) | 159.9 |
Table 2: AVITI delivers high-quality exome data
References
- Marian, A.J., “Sequencing Your Genome: What Does It Mean?” Methodist DeBakey Cardiovascular Journal 10, no. 1 (January 2014): 3–6, https://doi.org/10.14797/mdcj-10-1-3.
- Nurk, Sergey, Sergey Koren, Arang Rhie, et al., “The complete sequence of a human genome,” Science 376, no. 6588 (March 2022): 44–53, https://www.science.org/doi/10.1126/science.abj6987.
- Arslan, Sinan, Francisco J. Garcia, Minghao Guo, et al., “Sequencing by avidite enables high accuracy with low reagent consumption,” Nature Biotechnology (May 2023): https://doi.org/10.1038/s41587-023-01750-7.
- Carroll, Andrew, Alexey Kolesnikov, Daniel E. Cook, et al., “Accurate human genome analysis with Element Avidity sequencing,” bioRxiv (August 2023): https://doi.org/10.1101/2023.08.11.553043.
- “Picard,” Broad Institute, https://broadinstitute.github.io/picard/picard-metric-definitions.html.