Since Antonie van Leeuwenhoek first peered into a microscope to study microorganisms in the 17th century, microbiology has made the remarkably diverse, unseen world around us visible. The advent of next-generation sequencing (NGS) introduced a culture-free solution that revealed the true diversity of microbial communities living on, in, and around us, sparking deeper exploration into the composition and function of these important ecosystems.

Tools to assess microbes and their environments
A range of methods cultivate our understanding of the structure and function of microbial communities. The most comprehensive method is shotgun sequencing, which samples all genes in a community to understand its functional potential. Shotgun sequencing can answer questions about which fuel sources a community can consume, metabolites it can produce, and antibiotics it can resist. Metatranscriptomics enhances this assessment with a snapshot of ongoing activity in an environment at the time of sampling.
Together, metagenomics and metatranscriptomics form a formidable tool for understanding the influence a microbiome has on an environment. However, the throughput required for metagenome sequencing is often high, especially for soil and other complex communities. Ascertaining whether additional sequencing might pick up additional genes that are low abundance but functionally important is difficult and expensive.
An alternative, more economical approach is to focus on community composition via targeted sequencing. High-level community functions can be inferred from known information about the biology of member species. Moreover, the impacts of environmental perturbations or stratification are observable across many samples with far fewer sequencing reads. Even as sequencing costs have retreated, targeted sequencing remains the most popular metagenomics approach.
Targeted sequencing for metagenome profiling
The 16S rRNA gene is ideal for profiling composition. The gene is sufficiently conserved across bacterial clades for reliable amplification with universal primers, and with nine variable regions (V1–V9) contains enough sequence diversity to distinguish bacteria at the species or strain levels.
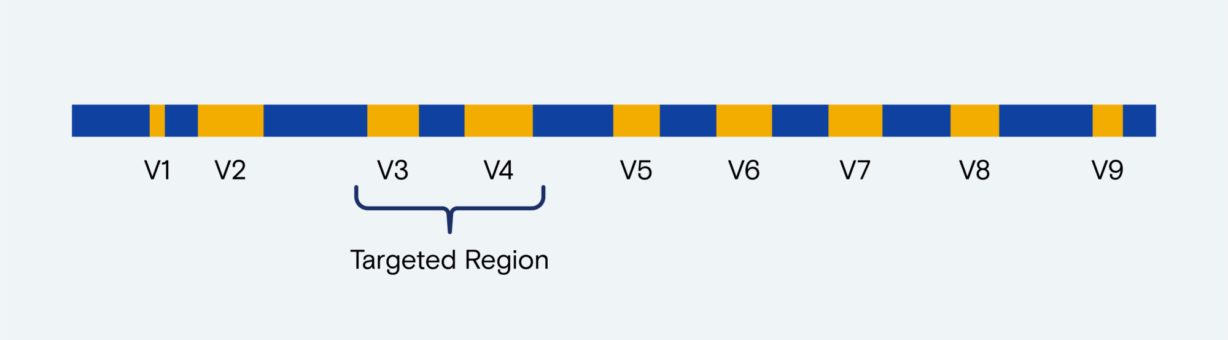
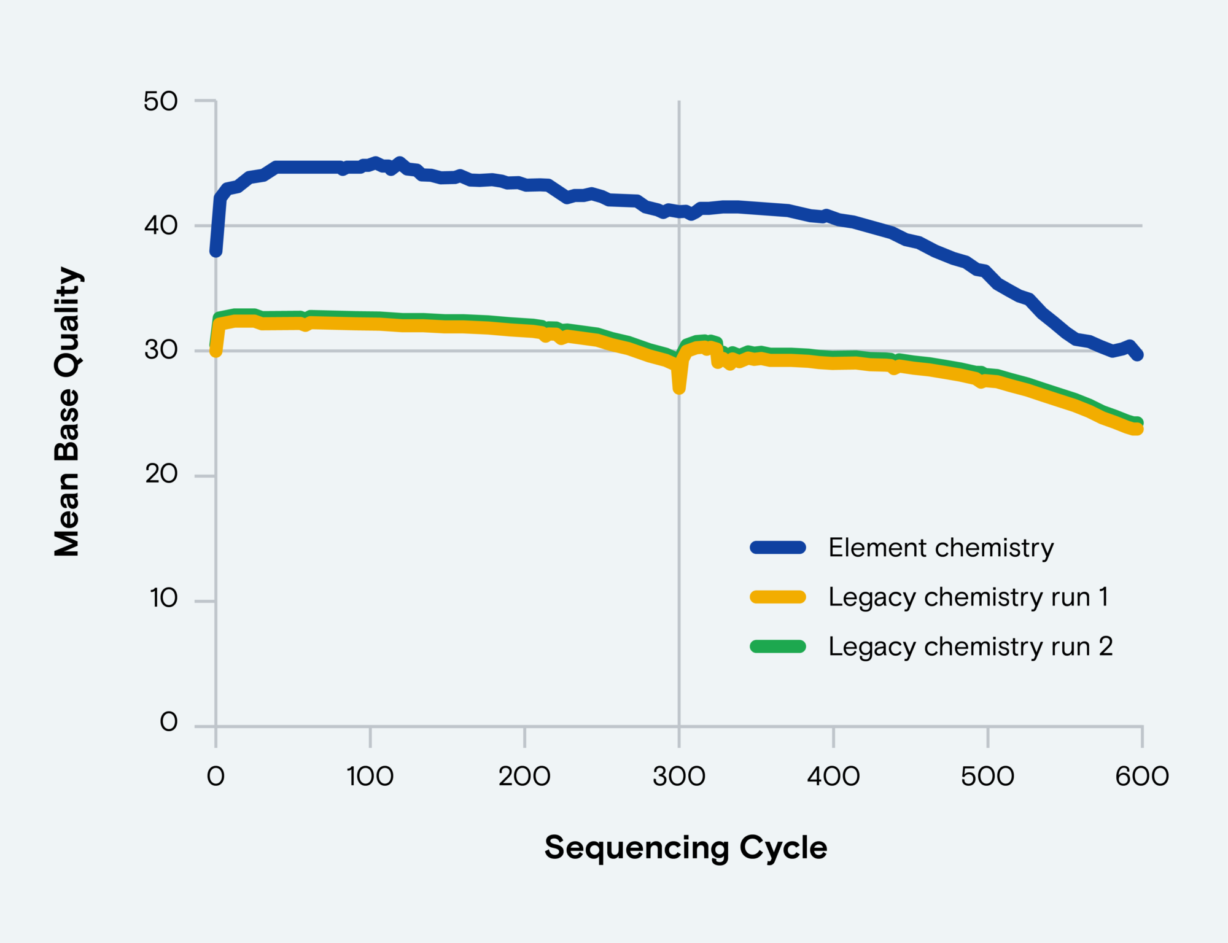
The sequence diversity of each variable region can differ between bacterial clades, so community profiling requires various amplified gene fragments. The common use of V3–V4 amplicons, which span longer gene fragments, requires up to 2 x 300 cycles of paired-end sequencing to produce a consensus sequence. At the ends of these long reads, sequencing-by-synthesis (SBS) accuracy declines, which can erode quality in the read overlap region.
Here, Element picks up where SBS falls short. Our chemistry generates 2 x 300 reads that maintain Q30–Q40+ quality throughout Read 1 and Read 2 to deliver high-confidence data for characterizing complex biological systems.