Cytosine methylation plays an important role in many biological processes, including cell lineage specification, X-chromosome inactivation, and the preservation of chromosome stability. Given its importance in so many basic cellular processes, errors in methylation have been linked to a wide range of human diseases, including cancer and autoimmune disease.
The detection of methylation patterns via DNA sequencing is an important tool for researchers trying to unwind the mechanisms of human disease and health, but it these experiments can be expensive due to the inherent low diversity of bisulfite samples and the limitations of traditional NGS technology.
The Challenges of Bisulfite Sequencing
To assess the methylation state of DNA via sequencing, it is common to convert unmethylated C’s to U’s either enzymatically or via bisulfite conversion. Following sequencing and an alignment to both a methylated and unmethylated reference, the methylated sites can be identified in the sample of interest.
However, because the vast majority of cytosines are unmethylated in most sample types, this leads to very few C base calls (and an overabundance of T base calls) in the resulting sequencing library. Libraries with one or more under-represented bases are termed low diversity and they pose a challenge for many sequencing technologies.
With many technologies, low diversity samples interfere with the ability to map the location of distinct clusters and maintain base calling accuracy as sequencing progresses. To overcome this challenge, it is common to either pool such libraries with high diversity libraries or to supplement the library with a significant PhiX DNA spike-in prior to sequencing. While amount of PhiX DNA required and the impact of low diversity on target density varies by sequencing platform, the addition of reduces the effective throughput of the run, driving up the cost of sequencing.
A Better Path to Low-Diversity Sequencing on the AVITI™ System
Unlike other sequencing platforms, the AVITI system does not require diversity to maintain accuracy as signals from the four bases are more reliably distinguishable due to our unique sequencing chemistry. In addition, AVITI libraries have specific characteristics that enable clean mapping of polonies during the initial cycles.
We decided to assess the capability of the AVITI Sequencing system on MethylSeq libraries. Our objective was to evaluate density and accuracy, while varying the PhiX spike-in percentage. Libraries were prepared using the NEBNext Enzymatic MethylSeq kits. Figure 1 shows the library preparation process.
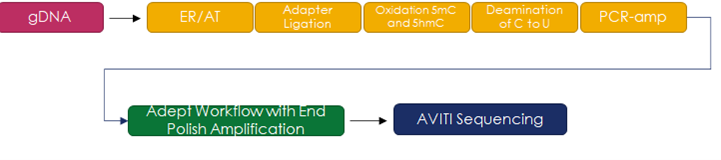
We sequenced the well-characterized sample NA12878 pooled with the addition of 1% each of a fully methylated control library (pUC19) and a fully unmethylated control library (phage lambda). Three runs were completed with a PhiX spike-in of 0%, 5%, and 20%, respectively by concentration. The summary of the primary sequencing metrics is provided in Table 1.
| Condition | PE Reads | %Q30 | PhiX Aligned (%) |
|---|---|---|---|
| No PhiX | 857 M | 94% | N/A |
| 5% PhiX | 920 M | 95% | 5.3% |
| 20% PhiX | 989 M | 96% | 27% |
Each run surpassed the 800M PE specification and attained a high percentage of Q30 bases. The PhiX spike-in for the third condition was higher than expected, reflecting either loading variation or some amplification preference for the PhiX reads.
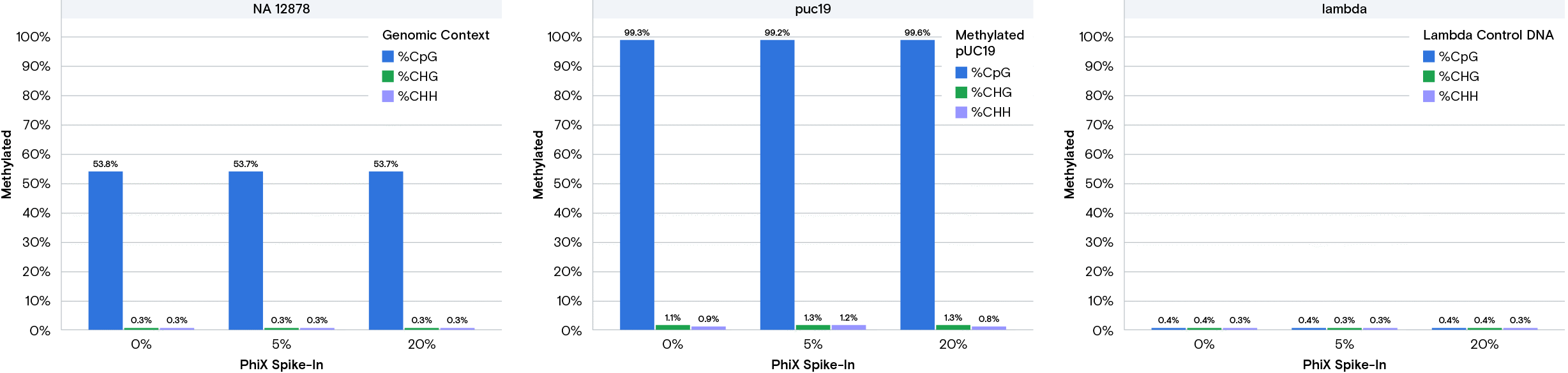
We processed the run using the NFCore implementation of the Bismark methylation pipeline to obtain the percentage of methylated CpG sites in each of the libraries. According to documentation from sample manufacturer NEB, the expected methylation percentage of CpG sites for the 3 libraries are 53% (NA112878), 100% (pUC19), and 0% (phage lambda). Figure 2 shows our results from the output of the Bismark pipeline.
Avoid the PhiX Tax to Lower Your Sequencing Cost
The methylation fraction closely matches the expected results and is highly consistent across runs even with very little PhiX present. The FASTQ data is publicly available on our website. These results show that the AVITI system is compatible with the NEBNext Enzymatic MethylSeq and produces high quality methylation data, even with no PhiX spike-in.
We still recommend a 5% PhiX spike-in for robustness and real-time error measurement.
However, a large amount of PhiX is not required to obtain accurate MethylSeq data on AVITI, further lowering the cost of sequencing relative to a competing mid-throughput platform by a further 15%, on top of the already lower cost of reagents.